Inleiding
Posterieur reversibel encefalopathiesyndroom (PRES) is een zeldzame complicatie van verschillende klinische entiteiten. De incidentie ervan is niet bekend en is gerapporteerd in een breed scala van leeftijden van 14 tot 78 jaar, met een gemiddelde leeftijd van 44 jaar en een man / vrouw-verhouding van 0,8 / 1,1. Hoewel de prognose doorgaans gunstig is, kunnen sterftecijfers tot 15 % zijn gemeld.2 Het wordt bepaald door typische klinisch-radiologische manifestaties, meestal van voorbijgaande aard.3 Ofwel in een acute of subacute vorm, in afnemende volgorde van frequentie treedt het op bij encefalopathie, toevallen, hoofdpijn, visuele stoornissen en focale neurologische uitval.4 Er zijn verschillende pathofysiologische theorieën gepostuleerd, waarvan er twee het meest worden geaccepteerd. De eerste suggereert dat de plotselinge stijging van de bloeddruk de zelfregulatie van de cerebrale bloedstroom overtreft, waardoor vasodilatatie en hyperperfusie ontstaat, met een breuk van de bloed-hersenbarrière (BBB) en vasogeen oedeem.5 Het wordt dus klassiek geassocieerd met eclampsie en hypertensieve encefalopathie; 20–30% van de patiënten is echter normotensief, wat een tweede theorie suggereert van directe endotheliale toxiciteit veroorzaakt door inflammatoire mediatoren, meer gecorreleerd met patiënten met immunosuppressieve behandeling, nierfalen, bindweefselaandoeningen of sepsis.6 Het komt voor in 7 Magnetische resonantiebeeldvorming ( MRI) van de hersenen is bepalend voor de diagnose en vertoont vasogeen oedeem, meestal in het achterste cerebrale gebied, bilateraal en symmetrisch.8 Aangezien de diagnose van PRES een hoge klinische en beeldvormende verdenking vereist, met de daaropvolgende vaststelling van een vroege behandeling voor een gunstige prognose; het doel van dit werk is om informatie te verschaffen voor de herkenning en behandeling van dit ongebruikelijke syndroom geassocieerd met SLE.
Beschrijving van de zaak
Een 25-jarige mestizo-vrouw, uit Quito, Ecuador, met persoonlijke pathologische antecedenten van hypothyreoïdie en SLE, gediagnosticeerd in december 2012, op de leeftijd van 21 jaar. In december 2016, 4 jaar na de diagnose van SLE, vertoonde ze een exacerbatie veroorzaakt door acute diarreeziekte; met musculoskeletale (artritis, myalgieën) en mucocutane (orale ulcera) manifestaties, serositis (rechter pleurale effusie), bicytopenie (anemie, trombocytopenie), nierbetrokkenheid (hematurie, proteïnurie, acuut nierfalen, acuut nierletsel Netwerkclassificatie III) en arteriële hypertensie (AHT); die haar allemaal een hoge SLEDAI opleverden (waarde: 23). De nierbiopsie meldde focale proliferatieve lupus glomerulonefritis klasse II, niet overeenkomend met intense lupusactiviteit; zonder veranderingen die kunnen worden toegeschreven aan het antifosfolipidensyndroom, evenals negativiteit van deze antilichamen. Vanwege de betrokkenheid van meerdere organen ontving ze pulsen van 1 g methylprednisolon intraveneus gedurende 3 dagen, vervanging van bloedproducten, 6 sessies plasmaferese, hemodyalisatie, amlodipine 10 mg / dag, atenolol 50 mg / dag en mycofenolaatmofetil 1 g / 12 uur oraal, sinds zij vertoonde gastro-intestinale intolerantie voor hogere doses. Drie weken na opname vertoonde ze een acceptabele analytische en klinische verbetering met de behandeling, en daarom was haar ontslag uit het ziekenhuis aangewezen. Vierentwintig uur later komt de patiënt terug met een krampachtige status. In noodgevallen begonnen ze met luchtwegbeheer, intraveneuze anticonvulsiva met diazepam 10 mg, midazolam 3 mg, fenytoïne 1 g en brachten haar over naar de intensive care.
Er werd een craniale tomografie aangevraagd, die geen tekenen van ischemie of bloeding vertoonde ; een rechter occipitale hypodensiteit zonder massa-effect werd waargenomen, en om deze reden werd de volgorde van onderzoeken verbreed om de etiologie ervan te identificeren. Metabole, infectieuze en farmacologische oorzaken werden uitgesloten. Omdat AHT moeilijk te beheersen is (tot 190/100 mmHg met een gemiddelde arteriële druk van 130 mmHg), had de patiënt tot 6 antihypertensiva nodig; atenolol 50 mg / 12 uur, losartan 100 mg / dag, amlodipine 10 mg / dag, doxazosine 2 mg / 6 uur via nasogastrische buis, en intraveneuze nitroprusside 50 mg / dag en furosemide 20 mg / 6 uur. Vanwege ernstige lupusactiviteit (SLEDAI 21: toevallen, hematurie, proteïnurie, hypocomplementemie, anti-DNA, trombocytopenie), kreeg ze opnieuw een behandeling met methylprednisolon 1 g / 3 dagen. Het elektro-encefalogram vertoonde geen epileptische activiteit. Er werd verzocht om een cerebrale angioresonantie waarbij geen bevindingen werden gevonden die consistent waren met vasculitis of trombose van het centrale zenuwstelsel. De MRI van de hersenen toonde typische beelden van PRES (Fig. 1), waarvan de ontwikkeling gerelateerd zou zijn aan de verergerde SLE, ernstige AHT, lupus glomerulonefritis en het gebruik van immunosuppressiva, dus oraal nimodipine 60 mg / 6 uur werd toegevoegd en de triggerfactoren werden gecontroleerd .Vanwege het risico op geneesmiddelgeïnduceerde lupus werd fenytoïne geleidelijk afgebouwd, met een progressieve toename van levetiracetam tot 1 g / 12 uur via een nasogastrische sonde. Tabel 1 geeft een overzicht van de relevante aanvullende onderzoeken. Tijdens de follow-up vertoonde ze geen nieuwe convulsies, de nierfunctie bleef stationair, de bloeddrukwaarden verbeterden (MAP 85-90 mmHg) en de lupusactiviteit nam af (SLEDAI 13: hematurie, proteïnurie, hypocomplementemie, anti-DNA trombocytopenie). De MRI van de controlerende hersenen toonde involutie van de vorige laesies (Fig. 2).
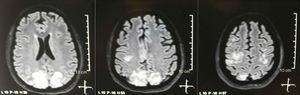
Simpele + diffusie nucleaire magnetische resonantie beeldvorming van de hersenen bij opname: bilaterale, symmetrische, hyperintense laesies in FLAIR sequentie in witte stof van occipitale en pariëtale lobben.
Relevante aanvullende studies van de casus.
| Datum | Klinische tests |
|---|---|
| Verergering van SLE 4 jaar na diagnose | HB: hemoglobine 7 mg / dl; hematocriet 22,2%; bloedplaatjes 75.000 / μlC-reactief proteïne: 2,52 mg / dl; procalcitonine 0,348 ng / ml Creatinine 5,7 mg / dl (eGFR CKD-EPI: 9,6 ml / min / 1,73 m2); ureum 187 mg / dl Creatinekinase: 18 U / l; lactaatdehydrogenase: 237 U / lMU: eiwitten 150 mg / dl; rode bloedcellen 40,8 / veld; proteïnurie: 1452 mg / 24h C3: 36 mg / dl; C4: 15 mg / dl; anti-dsDNA: 200 IE / ml Lupus-anticoagulans, anticardiolipinen, bèta-2-glycoproteïne 1: negatief Nierbiopsie; focale lupus proliferatieve glomerulonefritis klasse II (ISN / RPS) met een chroniciteits- en activiteitsindex van 4 |
| Heropname | HB: hemoglobine 8,90 mg / dl; hematocriet 26,3%; bloedplaatjes 130.000 / μl Creatinine 2,77 mg / dl (eGFR CKD-EPI: 22,8 ml / min / 1,73 m2); ureum 105,1 mg / dl Lactaatdehydrogenase: 527 E / l; y-glutamyltransferase 79U / l; alkalische fosfatase: 138 U / l; totaal bilirubine: 1,23 mg / dl; direct: 0,50 mg / dl; indirect: 0,73 mg / dlMU: eiwitten 300 mg / dl; rode bloedcellen 44 / veld; proteïnurie 1974 mg / 24 uur Eenvoudige craniale tomografie: rechter occipitale hypodensiteit, zonder massa-effect. Geen tekenen van ischemie of bloeding. Elektro-encefalogram: hersenactiviteit op de achtergrond bij 4 Hz in theta-ritme met laag voltage. Aanwezigheid van scherpe topgolven met posterieure deltaritmes met interhemisferische symmetrie, zonder paroxysmale epileptiforme activiteit Magnetische resonantie-angiografie van de hersenen in arteriële en veneuze fasen: zonder vasculaire veranderingen |
Anti-dsDNA: anti-dubbelstrengs DNA; HB: hematische biometrie; MU: microscopisch urineonderzoek; ISN / RPS: International Society of Nephrology / Renal Pathology Society; SLE: systemische lupus erythematosus; eGFR CKD-EPI: glomerulaire filtratiesnelheid geschat met behulp van de vergelijking van de Chronic Kidney Disease-Epidemiology Collaboration.
Discussie
Sinds de eerste beschrijving van de PRES, gemaakt in 1996 door Hinchey et al., is de kennis van verschillende aspecten van deze entiteit verbreed. De oorspronkelijke naam van het reversibele posterieure leuko-encefalopathiesyndroom was ongepast, aangezien de beeldvormingsveranderingen niet altijd beperkt zijn tot de witte hersenstof en de klinische manifestaties niet altijd omkeerbaar zijn.9 De eerste 15 gemelde gevallen traden op bij patiënten met hypertensieve encefalopathie, eclampsie of onder immunosuppressiva. behandeling.10 Het is ook waargenomen als een complicatie van andere entiteiten, zoals sepsis, nierfalen en bindweefselaandoeningen; daarom is het momenteel bekend dat de risicofactoren die endotheeldisfunctie veroorzaken de sleutel zijn voor de ontwikkeling van PRES.11 De wereldwijde incidentie is niet bekend, maar gegevens van retrospectieve onderzoeken geven aan dat het vaker voorkomt bij personen tussen 39 en 47 jaar. vrouwen met comorbiditeiten zoals hypertensieve, nier- of auto-immuunziekten.12 Bij patiënten met SLE zijn veel auto-antilichamen gericht tegen het endotheel; het produceren van zijn activering, expressie van adhesiemoleculen (E-selectine, VCAM-1, ICAM-1) en blootstelling aan pro-inflammatoire cytokines zoals IL-1β, TNFα en IL-6, waardoor de BBB wordt verstoord en neurologische complicaties optreden.13 Er is gerapporteerd dat bij mensen bij wie SLE is vastgesteld, de PRES optreedt in de context van matige tot ernstige lupusactiviteit, evenals in verband met nierfalen en slecht gecontroleerde hypertensie.14
Wat betreft de klinische manifestaties van de PRES, deze wordt gekenmerkt door variabele graden van encefalopathie, van verwarring tot verdoving (50-80%), toevallen (60-75%), hoofdpijn (50%) en visuele stoornissen variërend van wazig zien tot corticale blindheid (33%); ongebruikelijk zijn de focale neurologische uitval (10-15%) en de status epileptische (5-15%). 15 Voor de eerste beoordeling van het neurologische compromis bij deze patiënten wordt gewoonlijk een craniale computed axiale tomografie (CT) -scan gevraagd, die is vaak normaal of kan corticale-subcorticale hypodensiteiten vertonen, voornamelijk in de posterieure hersengebieden.16 De MRI van de hersenen bepaalt de diagnose en toont vasogeen oedeem, meestal in de witte stof van de occipitale en pariëtale lobben (gebied van de posterieure cerebrale circulatie) , gevisualiseerd als hyperintense laesies in T2 en FLAIR, bilateraal en symmetrisch.17 De preferentiële betrokkenheid van de witte stof is te wijten aan de structuur van gemyeliniseerde vezels, arteriolen en capillairen, waardoor het meer laks is. Evenzo kunnen de vaten van de voorste cerebrale circulatie, die een grotere sympathische innervatie hebben, adequaat reageren door vasoconstrictie op de plotselinge toename van de cerebrale bloedstroom secundair aan hypertensie; een beschermingsmechanisme dat minder ontwikkeld is in het vertebrobasilaire systeem.18 Minder vaak kunnen de grijze massa en andere lobben worden aangetast. De beelden met diffusiesequenties maken het mogelijk onderscheid te maken tussen het vasogene oedeem, typisch voor PRES, en het cytotoxische oedeem dat atypisch kan optreden en kan overgaan in een infarct.19 Het elektro-encefalogram correleert niet altijd met de neurologische aandoening, maar het kan encefalopathie aan het licht brengen door de aanwezigheid van focale scherpe golven. Bij patiënten met aanvallen die verband houden met PRES, is de belangrijkste elektro-encefalografische verandering de algemene vertraging van de theta / delta-frequenties.20 De analyse van het hersenvocht laat niet-specifieke veranderingen zien, zoals een lichte toename in cellulariteit en eiwitten, en daarom is het nuttig wanneer het handig om een infectie in het centrale zenuwstelsel uit te sluiten.21 Naast de bovengenoemde tests, degenen die noodzakelijk worden geacht voor de differentiële diagnose, voornamelijk met neurolupus, metabole en parainfectieuze encefalopathie, encefalitis, infarct van de a. cerebralis posterior en demyeliniserende aandoeningen moet worden uitgevoerd.22,23 Op basis van het voorgaande toont Fig. 3 het algoritme voorgesteld door Fugate et al. voor de diagnose van PRES, dat tot doel heeft zelfs atypische gevallen te identificeren.15
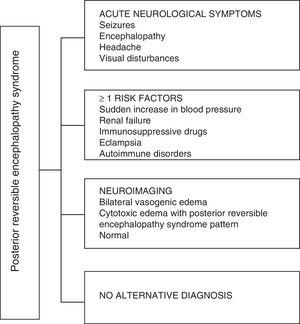
Diagnostisch algoritme voor posterieur reversibel encefalopathiesyndroom.
Gezien de klinische presentatie samen met de vele risicofactoren, beeldvormingsbevindingen en uitsluiting van andere etiologieën, was de diagnose PRES bij onze patiënt geconcludeerd. Symptomatische behandeling met anticonvulsieve medicatie en anti-cerebraal oedeem werd tijdig ingesteld, samen met de controle van de oorzakelijke factoren: ernstige hypertensie, SLE met ernstige activiteit, lupus glomerulonefritis, immunosuppressiva; bekrachtiging van de diagnose tijdens de follow-up met het oplossen van de klinische en beeldvormende wijzigingen. Met betrekking tot het beheer van de PRES moet de bloeddruk worden verlaagd, de aanvallen worden behandeld en moet de trigger worden gecontroleerd. De snelle verlaging van de bloeddruk kan cerebrale ischemie veroorzaken, en daarom wordt een doel van een gemiddelde bloeddruk tussen 105 en 125 mmHg voorgesteld, zonder 25% van deze verlaging in het eerste uur te overschrijden. De eerstelijnsgeneesmiddelen zijn calciumantagonisten (nicardipine of nimodipine naar keuze, dat ook cerebrale vasospasmen voorkomt) of bètablokkers (bijvoorbeeld labetalol). Natriumnitroprusside of hydralazine kunnen worden gebruikt als tweedelijnsgeneesmiddelen. Nitroglycerine moet worden vermeden vanwege het vaatverwijdende effect, dat het hersenoedeem zou versterken.24 De behandeling van de aanvallen is vergelijkbaar met die van andere epileptische aanvallen. Benzodiazepines zoals lorazepam of diazepam worden gebruikt als eerstelijnsbehandeling. Als tweedelijns, fenytoïne of valproaat, vooral bij status epilepticus, of fenobarbital. Magnesiumsulfaat kan worden gebruikt bij zwangere vrouwen. Bij refractaire aanvallen kunnen we propofol of pentobarbital geven.25 Geneesmiddelen die geneesmiddelgeïnduceerde lupus kunnen veroorzaken, zoals hydralazine, methyldopa, captopril, fenytoïne, valproaat en carbamazepine, moeten worden vermeden bij patiënten met SLE. Er is controverse over het beheer van immunosuppressiva bij de behandeling van PRES bij patiënten met SLE.26 Na het oplossen van de PRES komen epileptische aanvallen niet vaak voor, en daarom moet worden overwogen om anticonvulsiva te staken zolang er een adequate controle is de risicofactoren.27 Met een tijdige en adequate behandeling evolueert de meerderheid van de patiënten met PRES naar tevredenheid met remissie van de symptomen en beeldvormende laesies in een paar dagen of weken, hoewel complicaties, met name hemorragische, in 9-33% van de gevallen zijn waargenomen. casus benadrukt het belang van de herkenning en het beheer ervan, wat vaak een uitdaging is.28
Conclusies
De diagnose PRES vereist een hoog klinisch en beeldvormend vermoeden. Tijdige behandeling met controle van de symptomen en de onderliggende oorzaak bekrachtigt de diagnose tijdens de follow-up, met de oplossing van klinische en beeldvormende veranderingen; anders kan het neurologische gevolgen of de dood veroorzaken.
Belangenverstrengeling
De auteurs verklaren dat ze geen belangenverstrengeling hebben.