Einleitung
Das posteriore reversible Enzephalopathiesyndrom (PRES) ist eine seltene Komplikation verschiedener klinischer Einheiten. Die Inzidenz ist unbekannt und wurde in einem breiten Altersbereich von 14 bis 78 Jahren mit einem Durchschnittsalter von 44 Jahren und einem Verhältnis von Männern zu Frauen von 0,8 / 1,1 berichtet. Obwohl die Prognose normalerweise günstig ist, liegen die Sterblichkeitsraten bei bis zu 15 Jahren % wurden berichtet.2 Es wird durch typische klinisch-radiologische Manifestationen bestimmt, die normalerweise vorübergehend sind.3 Entweder in akuter oder subakuter Form, in absteigender Reihenfolge der Häufigkeit, tritt es bei Enzephalopathie, Anfällen, Kopfschmerzen, Sehstörungen und fokalem neurologischem Defizit auf.4 Es wurden mehrere pathophysiologische Theorien postuliert, von denen zwei am meisten akzeptiert werden. Der erste deutet darauf hin, dass der plötzliche Anstieg des Blutdrucks die Selbstregulation des zerebralen Blutflusses übersteigt und zu Vasodilatation und Hyperperfusion mit Bruch der Blut-Hirn-Schranke (BBB) und vasogenem Ödem führt.5 Daher wurde er klassisch mit assoziiert Eklampsie und hypertensive Enzephalopathie; 20–30% der Patienten sind jedoch normotensiv, was auf eine zweite Theorie der direkten endothelialen Toxizität hinweist, die durch Entzündungsmediatoren verursacht wird und stärker mit Patienten mit immunsuppressiver Behandlung, Nierenversagen, Bindegewebsstörungen oder Sepsis korreliert.6 Sie tritt in der 7-Magnetresonanztomographie auf ( Die MRT) des Gehirns ist für die Diagnose entscheidend und zeigt ein vasogenes Ödem, normalerweise im posterioren zerebralen Bereich, bilateral und symmetrisch.8 Da die Diagnose von PRES einen hohen klinischen und bildgebenden Verdacht erfordert, wird anschließend eine frühzeitige Behandlung für eine günstige Behandlung eingeleitet Prognose; Ziel dieser Arbeit ist es, Informationen zur Erkennung und Behandlung dieses ungewöhnlichen Syndroms im Zusammenhang mit SLE bereitzustellen.
Beschreibung des Falls
Eine 25-jährige Mestizenfrau aus Quito, Ecuador, mit persönlicher Pathologie Vorgeschichte von Hypothyreose und SLE, diagnostiziert im Dezember 2012, im Alter von 21 Jahren. Im Dezember 2016, 4 Jahre nach der Diagnose von SLE, stellte sie eine durch akute Durchfallerkrankung ausgelöste Exazerbation vor; mit muskuloskelettalen (Arthritis, Myalgien) und mukokutanen (orale Geschwüre) Manifestationen, Serositis (rechter Pleuraerguss), Bicytopenie (Anämie, Thrombozytopenie), Nierenbeteiligung (Hämaturie, Proteinurie, akutes Nierenversagen, akute Nierenverletzung Netzwerkklassifikation III) und arterieller Hypertonie (AHT); All dies verlieh ihr einen hohen SLEDAI (Wert: 23). Die Nierenbiopsie berichtete über fokale proliferative Lupusglomerulonephritis Klasse II, die nicht mit intensiver Lupusaktivität korrespondierte; ohne Veränderungen aufgrund des Antiphospholipid-Syndroms sowie der Negativität dieser Antikörper. Aufgrund der Beteiligung mehrerer Organe erhielt sie 3 Tage lang intravenös Impulse von 1 g Methylprednisolon intravenös, Ersatz von Blutprodukten, 6 Sitzungen Plasmapherese, Hämodyalisis, Amlodipin 10 mg / Tag, Atenolol 50 mg / Tag und Mycophenolatmofetil 1 g / 12 h oral Sie zeigte eine gastrointestinale Intoleranz gegenüber höheren Dosen. Drei Wochen nach der Aufnahme zeigte sie eine akzeptable analytische und klinische Verbesserung der Behandlung, weshalb ihre Entlassung aus dem Krankenhaus angezeigt war. 24 Stunden später tritt der Patient mit einem Krampfstatus wieder ein. In Notfällen leiteten sie ein Atemwegsmanagement ein, intravenöse Antikonvulsiva mit 10 mg Diazepam, 3 mg Midazolam und 1 g Phenytoin und brachten sie auf die Intensivstation.
Es wurde eine Schädeltomographie angefordert, die keine Anzeichen von Ischämie oder Blutung zeigte ;; Es wurde eine Hypodensität des rechten Hinterkopfes ohne Masseneffekt beobachtet, und aus diesem Grund wurde die Reihenfolge der Untersuchungen erweitert, um ihre Ätiologie zu identifizieren. Stoffwechsel-, infektiöse und pharmakologische Ursachen wurden ausgeschlossen. Aufgrund der schwer zu kontrollierenden AHT (bis zu 190/100 mmHg bei einem mittleren arteriellen Druck von 130 mmHg) benötigte der Patient bis zu 6 blutdrucksenkende Medikamente; Atenolol 50 mg / 12 h, Losartan 100 mg / Tag, Amlodipin 10 mg / Tag, Doxazosin 2 mg / 6 h über die Magensonde und intravenöses Nitroprussid 50 mg / Tag und Furosemid 20 mg / 6 h. Aufgrund schwerer Lupusaktivität (SLEDAI 21: Anfälle, Hämaturie, Proteinurie, Hypokomplementämie, Anti-DNA, Thrombozytopenie) erhielt sie erneut eine Behandlung mit Methylprednisolon 1 g / 3 Tage. Das Elektroenzephalogramm zeigte keine epileptiforme Aktivität. Es wurde eine zerebrale Angioresonanz angefordert, bei der keine Befunde im Zusammenhang mit Vaskulitis oder Thrombose des Zentralnervensystems gefunden wurden. Die MRT des Gehirns zeigte typische Bilder von PRES (1), deren Entwicklung mit dem verschlimmerten SLE, schwerer AHT, Lupusglomerulonephritis und der Verwendung von Immunsuppressiva zusammenhängen würde. Daher wurde orales Nimodipin 60 mg / 6 h hinzugefügt und die Triggerfaktoren wurden kontrolliert .Aufgrund des Risikos eines medikamenteninduzierten Lupus wurde Phenytoin schrittweise abgesetzt, wobei der Levetiracetam-Wert über die Magensonde schrittweise auf 1 g / 12 h anstieg. In Tabelle 1 sind die relevanten ergänzenden Studien aufgeführt. Während der Nachuntersuchung zeigte sie keine neuen Krampfereignisse, die Nierenfunktion blieb stationär, die Blutdruckwerte verbesserten sich (MAP 85–90 mmHg) und die Lupusaktivität nahm ab (SLEDAI 13: Hämaturie, Proteinurie, Hypokomplementämie, Anti-DNA) Thrombozytopenie). Die MRT des Kontrollgehirns zeigte eine Involution der vorherigen Läsionen (Abb. 2).
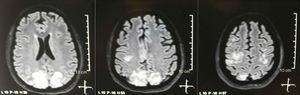
Einfache + Diffusions-Kernspinresonanztomographie des Gehirns bei Aufnahme: bilaterale, symmetrische, hyperintensive Läsionen in der FLAIR-Sequenz in der weißen Substanz der Okzipital- und Parietallappen.
Relevante ergänzende Studien zum Fall.
| Datum | Klinische Tests |
|---|---|
| Verschlimmerung von SLE 4 Jahre nach Diagnose | HB: Hämoglobin 7 mg / dl; Hämatokrit 22,2%; Blutplättchen 75.000 / μlC-reaktives Protein: 2,52 mg / dl; Procalcitonin 0,348 ng / ml Kreatinin 5,7 mg / dl (eGFR CKD-EPI: 9,6 ml / min / 1,73 m 2); Harnstoff 187 mg / dlKreatinkinase: 18 U / l; Lactatdehydrogenase: 237 U / lMU: Proteine 150 mg / dl; rote Blutkörperchen 40,8 / Feld; Proteinurie: 1452 mg / 24 hC3: 36 mg / dl; C4: 15 mg / dl; Anti-dsDNA: 200 IE / ml Lupus-Antikoagulans, Anticardiolipine, Beta-2-Glykoprotein 1: negativRenale Biopsie; fokale Lupus proliferative Glomerulonephritis Klasse II (ISN / RPS) mit einem Chronizitäts- und Aktivitätsindex von 4 |
| Rückübernahme | HB: Hämoglobin 8,90 mg / dl; Hämatokrit 26,3%; Blutplättchen 130.000 / μl Kreatinin 2,77 mg / dl (eGFR CKD-EPI: 22,8 ml / min / 1,73 m 2); Harnstoff 105,1 mg / dlLactatdehydrogenase: 527 U / l; γ-Glutamyltransferase 79U / l; alkalische Phosphatase: 138 U / l; Gesamtbilirubin: 1,23 mg / dl; direkt: 0,50 mg / dl; indirekt: 0,73 mg / dlMU: Proteine 300 mg / dl; rote Blutkörperchen 44 / Feld; Proteinurie 1974 mg / 24 h Einfache Schädeltomographie: Hypodensität des rechten Hinterkopfes ohne Masseneffekt. Keine Anzeichen von Ischämie oder Blutung Elektroenzephalogramm: Hintergrundgehirnaktivität bei 4 Hz im Niederspannungs-Theta-Rhythmus. Vorhandensein von scharfen Scheitelwellen mit posterioren Delta-Rhythmen mit interhemisphärischer Symmetrie, ohne paroxysmale epileptiforme AktivitätMagnetische Resonanzangiographie des Gehirns in arteriellen und venösen Phasen: ohne Gefäßveränderungen |
Anti-dsDNA: anti-doppelsträngige DNA; HB: hämatische Biometrie; MU: mikroskopische Urinanalyse; ISN / RPS: Internationale Gesellschaft für Nephrologie / Gesellschaft für Nierenpathologie; SLE: systemischer Lupus erythematodes; eGFR CKD-EPI: Glomeruläre Filtrationsrate, geschätzt unter Verwendung der Gleichung der Zusammenarbeit zwischen chronischer Nierenerkrankung und Epidemiologie.
Diskussion
Seit der Erstbeschreibung des PRES im Jahr 1996 durch Hinchey et al. wurde das Wissen über verschiedene Aspekte dieser Entität erweitert. Der ursprüngliche Name des reversiblen posterioren Leukoenzephalopathiesyndroms war unangemessen, da die bildgebenden Veränderungen nicht immer auf die zerebrale weiße Substanz beschränkt sind und die klinischen Manifestationen nicht immer reversibel sind.9 Die ersten 15 gemeldeten Fälle traten bei Patienten mit hypertensiver Enzephalopathie, Eklampsie oder unter Immunsuppressivität auf Behandlung.10 Es wurde auch als Komplikation anderer Entitäten wie Sepsis, Nierenversagen und Bindegewebsstörungen beobachtet; Daher ist derzeit bekannt, dass die Risikofaktoren, die eine endotheliale Dysfunktion verursachen, der Schlüssel für die Entwicklung von PRES sind.11 Die globale Inzidenz ist nicht bekannt, aber Daten aus retrospektiven Studien zeigen, dass sie bei Personen zwischen 39 und 47 Jahren im Allgemeinen häufiger auftritt Frauen mit Komorbiditäten wie Bluthochdruck-, Nieren- oder Autoimmunerkrankungen.12 Bei Patienten mit SLE sind viele Autoantikörper gegen das Endothel gerichtet; Aktivierung, Expression von Adhäsionsmolekülen (E-Selectin, VCAM-1, ICAM-1) und Exposition gegenüber proinflammatorischen Zytokinen wie IL-1β, TNFα und IL-6, was zu einer Störung der BHS und dem Auftreten neurologischer Komplikationen führt.13 Es wurde berichtet, dass bei Menschen, bei denen SLE diagnostiziert wurde, das PRES im Zusammenhang mit mittelschwerer bis schwerer Lupusaktivität sowie im Zusammenhang mit Nierenversagen und schlecht kontrollierter Hypertonie auftritt.14
In Bezug auf die klinischen Manifestationen des PRES ist es durch unterschiedliche Grade der Enzephalopathie gekennzeichnet, von Verwirrung bis Stupor (50–80%), Anfällen (60–75%), Kopfschmerzen (50%) und Sehstörungen von verschwommenem Sehen bis hin zu kortikaler Blindheit (33%); Ungewöhnlich ist das fokale neurologische Defizit (10–15%) und der Status epileptisch (5–15%). 15 Für die erste Beurteilung des neurologischen Kompromisses bei diesen Patienten wird normalerweise eine kraniale Computertomographie (CT) angefordert, die ist häufig normal oder kann kortikal-subkortikale Hypodensitäten aufweisen, vorwiegend in Regionen des hinteren Gehirns.16 Die MRT des Gehirns bestimmt die Diagnose und zeigt ein vasogenes Ödem, normalerweise in der weißen Substanz des Hinterhaupt- und Parietallappens (Gebiet des hinteren Gehirnkreislaufs). , dargestellt als hyperintensive Läsionen in T2 und FLAIR, bilateral und symmetrisch.17 Die bevorzugte Beteiligung der weißen Substanz beruht auf ihrer Struktur aus myelinisierten Fasern, Arteriolen und Kapillaren, die ihr eine größere Nachlässigkeit verleiht. In ähnlicher Weise können die Gefäße des vorderen Gehirnkreislaufs mit einer stärkeren sympathischen Innervation durch Vasokonstriktion angemessen auf den plötzlichen Anstieg des zerebralen Blutflusses infolge von Bluthochdruck reagieren; Ein im vertebrobasilaren System weniger entwickelter Schutzmechanismus.18 Weniger häufig können die graue Substanz und andere Lappen betroffen sein. Die Bilder mit Diffusionssequenzen ermöglichen die Unterscheidung zwischen dem für PRES typischen vasogenen Ödem und dem atypisch auftretenden zytotoxischen Ödem, das zum Infarkt führen kann.19 Das Elektroenzephalogramm korreliert nicht immer mit der neurologischen Beeinflussung, kann jedoch eine Enzephalopathie durch aufdecken das Vorhandensein von scharfen Wellen. Bei Patienten mit PRES-assoziierten Anfällen ist die hauptsächliche elektroenzephalographische Veränderung die allgemeine Verlangsamung der Theta / Delta-Frequenzen.20 Die Analyse der Cerebrospinalflüssigkeit zeigt unspezifische Veränderungen wie eine leichte Zunahme der Zellularität und der Proteine und ist daher nützlich, wenn dies der Fall ist Bequem, um eine Infektion des Zentralnervensystems auszuschließen.21 Zusätzlich zu den oben genannten Tests werden diejenigen, die für die Differentialdiagnose als notwendig erachtet werden, hauptsächlich mit Neurolupus, metabolischer und parainfektiöser Enzephalopathie, Enzephalitis, Infarkt der hinteren Hirnarterie und demyelinisierenden Störungen muss durchgeführt werden.22,23 Auf der Grundlage des Vorstehenden zeigt Fig. 3 den von Fugate et al. für die Diagnose von PRES, mit der auch atypische Fälle identifiziert werden sollen.15
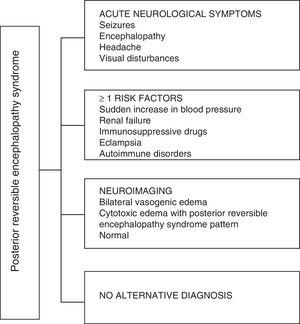
Diagnosealgorithmus für das posteriore reversible Enzephalopathiesyndrom.
Bei unserem Patienten war die Diagnose von PRES angesichts des klinischen Erscheinungsbilds zusammen mit den zahlreichen Risikofaktoren, den bildgebenden Befunden und dem Ausschluss anderer Ätiologien abgeschlossen. Die symptomatische Behandlung mit Antikonvulsiva und Antizerebralödemen wurde zeitnah eingeleitet, zusammen mit der Kontrolle der ursächlichen Faktoren: schwerer Bluthochdruck, SLE mit schwerer Aktivität, Lupus-Glomerulonephritis, Immunsuppressiva; Bestätigung der Diagnose während des Follow-up mit der Auflösung der klinischen und bildgebenden Veränderungen. In Bezug auf das Management des PRES sollte der Blutdruck gesenkt, die Anfälle behandelt und der Auslöser kontrolliert werden. Der rasche Blutdruckabfall kann zu zerebraler Ischämie führen, weshalb ein Ziel eines mittleren Blutdrucks zwischen 105 und 125 mmHg vorgeschlagen wird, ohne 25% dieser Blutdrucksenkung in der ersten Stunde zu überschreiten. Die Medikamente der ersten Wahl sind Kalziumkanalblocker (Nicardipin oder Nimodipin der Wahl, die auch einen zerebralen Vasospasmus verhindern) oder Betablocker (z. B. Labetalol). Natriumnitroprussid oder Hydralazin können als Arzneimittel der zweiten Wahl verwendet werden. Nitroglycerin sollte aufgrund seiner vasodilatatorischen Wirkung, die das Hirnödem verstärken würde, vermieden werden.24 Die Behandlung der Anfälle ähnelt der anderer epileptischer Anfälle. Benzodiazepine wie Lorazepam oder Diazepam werden als Erstlinientherapie eingesetzt. Als zweite Linie Phenytoin oder Valproat, insbesondere bei Status epilepticus oder Phenobarbital. Magnesiumsulfat kann bei schwangeren Frauen angewendet werden. Bei refraktären Anfällen können wir Propofol oder Pentobarbital verabreichen.25 Bei Patienten mit SLE sollten Arzneimittel vermieden werden, die medikamenteninduzierten Lupus verursachen können, wie Hydralazin, Methyldopa, Captopril, Phenytoin, Valproat und Carbamazepin. Es gibt Kontroversen über die Behandlung von Immunsuppressiva bei der Behandlung von PRES bei Patienten mit SLE.26 Nach der Auflösung des PRES sind Anfälle selten, und daher sollte erwogen werden, Antikonvulsiva abzusetzen, solange eine angemessene Kontrolle über PRES besteht die Risikofaktoren.27 Bei rechtzeitiger und angemessener Behandlung entwickelt sich die Mehrheit der Patienten mit PRES zufriedenstellend mit einer Remission der Symptome und bildgebenden Läsionen innerhalb weniger Tage oder Wochen, obwohl in 9–33% der Fälle Komplikationen, insbesondere hämorrhagische, beobachtet wurden Der Fall unterstreicht die Bedeutung seiner Erkennung und Behandlung, die häufig eine Herausforderung darstellt.28
Schlussfolgerungen
Die Diagnose von PRES erfordert einen hohen klinischen und bildgebenden Verdacht. Eine rechtzeitige Behandlung mit Kontrolle der Symptome und der zugrunde liegenden Ursache bestätigt die Diagnose während der Nachsorge, wobei klinische und bildgebende Veränderungen behoben werden. Andernfalls kann es zu neurologischen Folgen oder zum Tod kommen.
Interessenkonflikt
Die Autoren erklären, dass sie keinen Interessenkonflikt haben.