Introduction
Le syndrome dencéphalopathie postérieure réversible (PRES) est une complication rare de diverses entités cliniques. Son incidence est inconnue, ayant été rapportée dans une large gamme dâges de 14 à 78 ans, avec un âge moyen de 44 ans et un rapport homme / femme de 0,8 / 1,1 Bien que le pronostic soit généralement favorable, des taux de mortalité allant jusquà 15 % ont été rapportés2. Il est déterminé par des manifestations cliniques-radiologiques typiques, généralement transitoires3. Sous une forme aiguë ou subaiguë, par ordre décroissant de fréquence, il se produit avec une encéphalopathie, des convulsions, des maux de tête, des troubles visuels et un déficit neurologique focal.4 Plusieurs théories physiopathologiques ont été postulées, deux étant les plus acceptées. Le premier suggère que laugmentation soudaine de la pression artérielle dépasse lautorégulation du flux sanguin cérébral, provoquant une vasodilatation et une hyperperfusion, avec rupture de la barrière hémato-encéphalique (BHE) et œdème vasogénique.5 Ainsi, elle a été classiquement associée à éclampsie et encéphalopathie hypertensive; cependant, 20 à 30% des patients sont normotendus, ce qui suggère une deuxième théorie de la toxicité endothéliale directe causée par des médiateurs inflammatoires, plus corrélée aux patients sous traitement immunosuppresseur, insuffisance rénale, troubles du tissu conjonctif ou septicémie.6 Elle se produit en 7 imagerie par résonance magnétique ( IRM) du cerveau est déterminante pour le diagnostic, montrant un œdème vasogénique, généralement dans le territoire cérébral postérieur, bilatéral et symétrique.8 Le diagnostic de PRES nécessite une forte suspicion clinique et dimagerie, avec la mise en place ultérieure dun traitement précoce pour une pronostic; lobjectif de ce travail est de fournir des informations pour la reconnaissance et la prise en charge de ce syndrome inhabituel associé au LED.
Description du cas
Une métisse de 25 ans, de Quito, Equateur, avec une pathologie personnelle antécédents dhypothyroïdie et de LED, diagnostiqués en décembre 2012, à lâge de 21 ans. En décembre 2016, 4 ans après le diagnostic de LED, elle a présenté une exacerbation provoquée par une maladie diarrhéique aiguë; avec manifestations musculo-squelettiques (arthrite, myalgies) et mucocutanées (ulcères buccaux), sérosite (épanchement pleural droit), bicytopénie (anémie, thrombocytopénie), atteinte rénale (hématurie, protéinurie, insuffisance rénale aiguë, classification III du Réseau des lésions rénales aiguës) et hypertension artérielle (AHT); tout cela lui a conféré un SLEDAI élevé (valeur: 23). La biopsie rénale a rapporté une glomérulonéphrite lupique proliférative focale de classe II, ne correspondant pas à une activité lupique intense; sans changements attribuables au syndrome des antiphospholipides, ainsi quà la négativité de ces anticorps. En raison de limplication de plusieurs organes, elle a reçu des impulsions de 1 g de méthylprednisolone par voie intraveineuse pendant 3 jours, remplacement des produits sanguins, 6 séances de plasmaphérèse, hémodyalisation, amlodipine 10 mg / jour, aténolol 50 mg / jour et mycophénolate mofétil 1 g / 12 h par voie orale, depuis elle a présenté une intolérance gastro-intestinale à des doses plus élevées. Trois semaines après son admission, elle a présenté une amélioration analytique et clinique acceptable avec le traitement et, par conséquent, sa sortie de lhôpital a été indiquée. Vingt-quatre heures plus tard, le patient rentre avec un état convulsif. En cas durgence, ils ont initié une prise en charge des voies respiratoires, des anticonvulsivants intraveineux avec du diazépam 10 mg, du midazolam 3 mg, de la phénytoïne 1 g et lont transférée à lunité de soins intensifs.
Il a été demandé une tomographie crânienne, qui na pas montré de signes dischémie ou de saignement ; une hypodensité occipitale droite sans effet de masse a été observée, et pour cette raison lordre des examens a été élargi pour identifier son étiologie. Les causes métaboliques, infectieuses et pharmacologiques ont été exclues. En raison de lAHT difficile à contrôler (jusquà 190/100 mmHg avec une pression artérielle moyenne de 130 mmHg), le patient a eu besoin de jusquà 6 antihypertenseurs; aténolol 50 mg / 12 h, losartan 100 mg / jour, amlodipine 10 mg / jour, doxazosine 2 mg / 6 h par sonde nasogastrique et nitroprussiate intraveineuse 50 mg / jour et furosémide 20 mg / 6 h. En raison dune activité lupique sévère (SLEDAI 21: convulsions, hématurie, protéinurie, hypocomplémentémie, anti-ADN, thrombocytopénie), elle a de nouveau reçu un traitement par méthylprednisolone 1g / 3 jours. Lélectroencéphalogramme na pas montré dactivité épileptiforme. Il a été demandé une angiorésonance cérébrale dans laquelle des résultats compatibles avec une vascularite ou une thrombose du système nerveux central nont pas été trouvés. LIRM du cerveau a montré des images typiques de PRES (Fig.1), dont le développement serait lié au SLE exacerbé, AHT sévère, glomérulonéphrite lupique et à lutilisation dimmunosuppresseurs, donc la nimodipine orale 60 mg / 6h a été ajoutée et les facteurs déclenchants ont été contrôlés .En raison du risque de lupus dorigine médicamenteuse, la phénytoïne a été progressivement supprimée, avec une augmentation progressive du lévétiracétam jusquà 1 g / 12 h par sonde nasogastrique. Le tableau 1 détaille les études complémentaires pertinentes. Au cours du suivi, elle na pas présenté de nouveaux événements convulsifs, la fonction rénale est restée stationnaire, les valeurs de la pression artérielle se sont améliorées (MAP 85–90 mmHg) et lactivité lupique a diminué (SLEDAI 13: hématurie, protéinurie, hypocomplémentémie, anti-ADN , thrombocytopénie). LIRM du cerveau de contrôle a mis en évidence linvolution des lésions précédentes (Fig. 2).
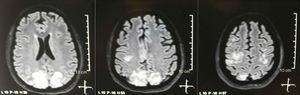
Imagerie par résonance magnétique nucléaire simple + diffusion du cerveau à ladmission: lésions bilatérales, symétriques, hyperintenses en séquence FLAIR dans la substance blanche des lobes occipitaux et pariétaux.
Études complémentaires pertinentes du cas.
| Date | Tests cliniques |
|---|---|
| Exacerbation du LED 4 ans après diagnostic | HB: hémoglobine 7 mg / dl; hématocrite 22,2%; plaquettes 75 000 / μlC-protéine réactive: 2,52 mg / dl; procalcitonine 0,348 ng / ml Créatinine 5,7 mg / dl (eGFR CKD-EPI: 9,6 ml / min / 1,73 m2); urée 187 mg / dl Créatine kinase: 18 U / l; lactate déshydrogénase: 237U / lMU: protéines 150 mg / dl; globules rouges 40,8 / champ; protéinurie: 1452 mg / 24hC3: 36 mg / dl; C4: 15 mg / dl; anti-ADNdb: 200 UI / ml Anticoagulant lupus, anticardiolipines, bêta 2-glycoprotéine 1: négatif Biopsie rénale; glomérulonéphrite proliférative lupique focale de classe II (ISN / RPS) avec un indice de chronicité et dactivité de 4 |
| Réadmission | HB: hémoglobine 8,90 mg / dl; hématocrite 26,3%; plaquettes 130 000 / μl Créatinine 2,77 mg / dl (eGFR CKD-EPI: 22,8 ml / min / 1,73 m2); urée 105,1 mg / dl lactate déshydrogénase: 527 U / l; γ-glutamyl transférase 79U / l; phosphatase alcaline: 138U / l; bilirubine totale: 1,23 mg / dl; direct: 0,50 mg / dl; indirect: 0,73 mg / dlMU: protéines 300 mg / dl; globules rouges 44 / champ; protéinurie 1974mg / 24hTomographie crânienne simple: hypodensité occipitale droite, sans effet de masse. Aucun signe dischémie ou de saignement Electroencéphalogramme: activité cérébrale de fond à 4 Hz en rythme thêta basse tension. Présence dondes aiguës au sommet avec des rythmes delta postérieurs à symétrie interhémisphérique, sans activité épileptiforme paroxystique Angiographie par résonance magnétique du cerveau en phases artérielle et veineuse: sans altérations vasculaires |
Anti-ADNdb: ADN anti-double brin; HB: biométrie hématique; MU: analyse durine microscopique; ISN / RPS: Société internationale de néphrologie / Société de pathologie rénale; LED: lupus érythémateux disséminé; eGFR CKD-EPI: taux de filtration glomérulaire estimé à laide de léquation de la Collaboration Maladie Rénale Chronique-Epidémiologie.
Discussion
Depuis la première description du PRES, faite en 1996 par Hinchey et al., la connaissance de plusieurs aspects de cette entité sest élargie. Son nom dorigine de syndrome de leucoencéphalopathie postérieure réversible sest avéré inapproprié, car les changements dimagerie ne se limitent pas toujours à la substance blanche cérébrale et ses manifestations cliniques ne sont pas toujours réversibles.9 Les 15 premiers cas rapportés sont survenus chez des patients atteints dencéphalopathie hypertensive, déclampsie ou sous immunosuppresseur. traitement.10 Il a également été observé comme une complication dautres entités telles que la septicémie, linsuffisance rénale et les troubles du tissu conjonctif; par conséquent, on sait actuellement que les facteurs de risque à lorigine dun dysfonctionnement endothélial sont essentiels pour le développement du PRES.11 Lincidence globale nest pas connue, mais les données détudes rétrospectives indiquent quelle est plus fréquente chez les individus entre 39 et 47 ans, généralement les femmes présentant des comorbidités telles que des troubles hypertensifs, rénaux ou auto-immunes.12 Chez les patients atteints de LED, de nombreux autoanticorps sont dirigés contre lendothélium; produisant son activation, lexpression de molécules dadhésion (E-sélectine, VCAM-1, ICAM-1) et lexposition à des cytokines pro-inflammatoires telles que lIL-1β, le TNFα et lIL-6, provoquant une perturbation du BBB et lapparition de complications neurologiques.13 Il a été rapporté que chez les personnes diagnostiquées avec un LED, le SEPR survient dans le contexte dune activité lupique modérée à sévère, ainsi que associé à une insuffisance rénale et une hypertension mal contrôlée.14
Concernant les manifestations cliniques du PRES, il est caractérisé par des degrés variables dencéphalopathie, de la confusion à la stupeur (50–80%), des convulsions (60–75%), des maux de tête (50%) et troubles visuels allant de la vision trouble à la cécité corticale (33%); étant inhabituel le déficit neurologique focal (10-15%) et létat de mal épileptique (5-15%). 15 Pour lévaluation initiale du compromis neurologique chez ces patients, une tomodensitométrie axiale (TDM) crânienne est généralement demandée, qui est souvent normale ou peut présenter des hypodensités cortico-sous-corticales, principalement dans les régions postérieures du cerveau.16 LIRM du cerveau détermine le diagnostic, montrant un œdème vasogénique, généralement dans la substance blanche des lobes occipital et pariétal (territoire de la circulation cérébrale postérieure) , visualisées comme des lésions hyperintenses en T2 et FLAIR, bilatérales et symétriques.17 Latteinte préférentielle de la substance blanche est due à sa structure de fibres myélinisées, artérioles et capillaires qui lui confère une plus grande laxité. De même, les vaisseaux de la circulation cérébrale antérieure, ayant une plus grande innervation sympathique, peuvent répondre adéquatement par vasoconstriction à laugmentation soudaine du flux sanguin cérébral secondaire à lhypertension; un mécanisme de protection moins développé dans le système vertébrobasilaire.18 Moins fréquemment, la matière grise et les autres lobes peuvent être affectés. Les images avec séquences de diffusion permettent de distinguer lœdème vasogène, typique du PRES, et lœdème cytotoxique pouvant survenir de manière atypique et évoluer vers un infarctus.19 Lélectroencéphalogramme nest pas toujours corrélé à laffectation neurologique, mais il peut révéler une encéphalopathie par la présence dondes focales nettes. Chez les patients présentant des crises associées au PRES, la principale altération électroencéphalographique est le ralentissement général des fréquences thêta / delta.20 Lanalyse du liquide céphalo-rachidien montre des changements non spécifiques tels quune légère augmentation de la cellularité et des protéines, et par conséquent elle est utile lorsquelle est pratique pour écarter une infection du système nerveux central.21 Outre les tests susmentionnés, ceux qui sont considérés comme nécessaires pour le diagnostic différentiel, principalement avec neurolupus, encéphalopathie métabolique et parainfectieuse, encéphalite, infarctus de lartère cérébrale postérieure et troubles démyélinisants doit être effectuée.22,23 Sur la base de ce qui précède, la figure 3 montre lalgorithme proposé par Fugate et al. pour le diagnostic de PRES, qui vise à identifier même les cas atypiques.15
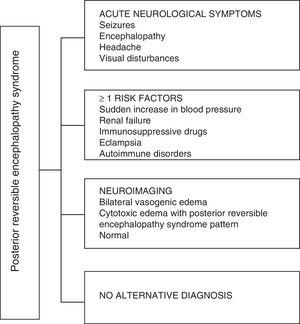
Algorithme de diagnostic du syndrome dencéphalopathie postérieure réversible.
Chez notre patient, compte tenu de la présentation clinique ainsi que des multiples facteurs de risque, des résultats dimagerie et de lexclusion dautres étiologies, le diagnostic de PRES était conclu. Un traitement symptomatique avec des médicaments anticonvulsivants et un œdème anti-cérébral a été instauré en temps opportun, ainsi que le contrôle des facteurs causaux: hypertension sévère, LED avec activité sévère, glomérulonéphrite lupique, médicaments immunosuppresseurs; la ratification du diagnostic lors du suivi avec la résolution des altérations cliniques et dimagerie. En ce qui concerne la gestion du PRES, la pression artérielle doit être réduite, les crises doivent être traitées et le déclencheur doit être contrôlé. La baisse rapide de la pression artérielle pourrait provoquer une ischémie cérébrale, cest pourquoi un objectif dune pression artérielle moyenne entre 105 et 125 mmHg est suggéré, sans dépasser 25% de cette réduction dans la première heure. Les médicaments de première intention sont des inhibiteurs calciques (nicardipine ou nimodipine de choix, qui prévient également le vasospasme cérébral) ou des bêtabloquants (par exemple, le labétalol). Le nitroprussiate de sodium ou lhydralazine peuvent être utilisés comme médicaments de deuxième intention. La nitroglycérine doit être évitée en raison de son effet vasodilatateur, qui augmenterait lœdème cérébral.24 Le traitement des crises est similaire à celui des autres crises dépilepsie. Les benzodiazépines telles que le lorazépam ou le diazépam sont utilisées comme traitement de première intention. En deuxième intention, phénytoïne ou valproate, notamment en état de mal épileptique, ou phénobarbital. Le sulfate de magnésium peut être utilisé chez la femme enceinte. Dans les crises réfractaires, nous pouvons administrer du propofol o pentobarbital.25 Les médicaments susceptibles de provoquer un lupus dorigine médicamenteuse, tels que lhydralazine, la méthyldopa, le captopril, la phénytoïne, le valproate et la carbamazépine, doivent être évités chez les patients atteints de LED. Il existe une controverse sur la gestion des médicaments immunosuppresseurs dans le traitement du SEPR chez les patients atteints de LED.26 Après la résolution du SEPR, les crises sont peu fréquentes et, par conséquent, il faut envisager darrêter les anticonvulsivants tant quil existe un contrôle adéquat des les facteurs de risque.27 Avec un traitement opportun et adéquat, la majorité des patients atteints de SEPR évoluent de manière satisfaisante avec une rémission des symptômes et des lésions dimagerie en quelques jours ou semaines, bien que des complications, particulièrement hémorragiques, aient été observées dans 9 à 33% des cas, Le cas met en évidence limportance de sa reconnaissance et de sa prise en charge, ce qui est souvent un défi.28
Conclusions
Le diagnostic de PRES nécessite une forte suspicion clinique et dimagerie. Un traitement rapide avec contrôle des symptômes et de la cause sous-jacente ratifie le diagnostic lors du suivi, avec la résolution des altérations cliniques et dimagerie; sinon, cela peut entraîner des séquelles neurologiques ou la mort.
Conflit dintérêts
Les auteurs déclarent ne pas avoir de conflit dintérêts.