Introducción
El síndrome de encefalopatía posterior reversible (SEPR) es una complicación poco frecuente de diversas entidades clínicas. Se desconoce su incidencia, habiéndose reportado en un amplio rango de edades desde los 14 a los 78 años, con una edad promedio de 44 años y una razón hombre / mujer de 0,8 / 1,1. Aunque el pronóstico suele ser favorable, tasas de mortalidad de hasta 15 años. Se han reportado %2. Está determinada por las manifestaciones clínico-radiológicas típicas, generalmente transitorias3. Bien en forma aguda o subaguda, en orden descendente de frecuencia se presenta con encefalopatía, convulsiones, cefalea, alteraciones visuales y déficit neurológico focal4. Se han postulado varias teorías fisiopatológicas, siendo dos las más aceptadas. El primero sugiere que el aumento brusco de la presión arterial supera la autorregulación del flujo sanguíneo cerebral, provocando vasodilatación e hiperperfusión, con ruptura de la barrera hematoencefálica (BHE) y edema vasogénico5. Así, clásicamente se ha asociado con eclampsia y encefalopatía hipertensiva; sin embargo, el 20-30% de los pacientes son normotensos, lo que sugiere una segunda teoría de toxicidad endotelial directa causada por mediadores inflamatorios, más correlacionada con pacientes con tratamiento inmunosupresor, insuficiencia renal, trastornos del tejido conjuntivo o sepsis.6 Se presenta en 7 Imágenes por resonancia magnética ( La resonancia magnética cerebral) es determinante para el diagnóstico, mostrando edema vasogénico, generalmente en el territorio cerebral posterior, bilateral y simétrico8. Dado que el diagnóstico de SEPR requiere una alta sospecha clínica e imagenológica, con el posterior establecimiento de un tratamiento precoz por una evolución favorable pronóstico; El objetivo de este trabajo es brindar información para el reconocimiento y manejo de este insólito síndrome asociado al LES.
Descripción del caso
Mujer mestiza de 25 años, de Quito, Ecuador, con patología personal antecedentes de hipotiroidismo y LES, diagnosticado en diciembre de 2012, a la edad de 21 años. En diciembre de 2016, 4 años después del diagnóstico de LES, presentó una exacerbación desencadenada por enfermedad diarreica aguda; con manifestaciones musculoesqueléticas (artritis, mialgias) y mucocutáneas (úlceras orales), serositis (derrame pleural derecho), bicitopenia (anemia, trombocitopenia), afectación renal (hematuria, proteinuria, insuficiencia renal aguda, Acute Kidney Injury Network clasificación III) e hipertensión arterial (AHT); todo lo cual le confirió un alto SLEDAI (valor: 23). La biopsia renal reportó glomerulonefritis lúpica proliferativa focal clase II, no correspondiendo con actividad lúpica intensa; sin cambios atribuibles al síndrome antifosfolípido, así como negatividad de estos anticuerpos. Debido a la afectación multiorgánica, recibió pulsos de 1g de metilprednisolona intravenosa durante 3 días, reposición de hemoderivados, 6 sesiones de plasmaféresis, hemodiálisis, amlodipino 10 mg / día, atenolol 50 mg / día y micofenolato mofetilo 1g / 12h vía oral, ya que presentó intolerancia gastrointestinal a dosis más altas. A las tres semanas del ingreso presentó una aceptable mejoría analítica y clínica con el tratamiento, por lo que se indicó su alta hospitalaria. Veinticuatro horas después, el paciente reingresa con un estado convulsivo. En urgencias iniciaron manejo de vía aérea, anticonvulsivantes intravenosos con diazepam 10mg, midazolam 3mg, fenitoína 1g y la trasladaron a la unidad de cuidados intensivos.
Se solicitó tomografía craneal, que no mostró signos de isquemia ni sangrado. ; Se observó una hipodensidad occipital derecha sin efecto de masa, por lo que se amplió el orden de exámenes para identificar su etiología. Se excluyeron las causas metabólicas, infecciosas y farmacológicas. Debido a la dificultad para controlar la HTA (hasta 190/100 mmHg con una presión arterial media de 130 mmHg), el paciente requirió hasta 6 fármacos antihipertensivos; atenolol 50 mg / 12 h, losartán 100 mg / día, amlodipino 10 mg / día, doxazosina 2 mg / 6 h vía sonda nasogástrica y nitroprusiato intravenoso 50 mg / día y furosemida 20 mg / 6 h. Debido a la actividad lúpica severa (SLEDAI 21: convulsiones, hematuria, proteinuria, hipocomplementemia, anti-ADN, trombocitopenia), recibió nuevamente tratamiento con metilprednisolona 1g / 3 días. El electroencefalograma no mostró actividad epileptiforme. Se solicitó una angiorresonancia cerebral en la que no se encontraron hallazgos compatibles con vasculitis o trombosis del sistema nervioso central. La RM de cerebro mostró imágenes típicas de PRES (fig.1), cuyo desarrollo estaría relacionado con el LES exacerbado, HTA severa, glomerulonefritis lúpica y uso de inmunosupresores, por lo que se añadió nimodipino oral 60mg / 6h y se controlaron los factores desencadenantes .Debido al riesgo de lupus inducido por fármacos, la fenitoína se retiró gradualmente, con aumento progresivo de levetiracetam hasta 1 g / 12 h por sonda nasogástrica. La Tabla 1 detalla los estudios complementarios relevantes. Durante el seguimiento no presentó nuevos eventos convulsivos, la función renal se mantuvo estacionaria, los valores de presión arterial mejoraron (PAM 85-90mmHg) y la actividad lúpica disminuyó (SLEDAI 13: hematuria, proteinuria, hipocomplementemia, anti-ADN , trombocitopenia). La resonancia magnética del cerebro de control evidenció involución de las lesiones previas (Fig. 2).
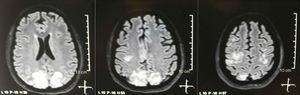
Resonancia magnética nuclear simple + difusión del cerebro al ingreso: lesiones bilaterales, simétricas, hiperintensas en secuencia FLAIR en la sustancia blanca de los lóbulos occipital y parietal.
Estudios complementarios relevantes del caso.
| Fecha | Pruebas clínicas |
|---|---|
| Exacerbación del LES 4 años después del diagnóstico | HB: hemoglobina 7 mg / dl; hematocrito 22,2%; plaquetas 75.000 / μl proteína reactiva C: 2,52 mg / dl; procalcitonina 0,348 ng / ml Creatinina 5,7 mg / dl (eGFR CKD-EPI: 9,6 ml / min / 1,73 m2); urea 187 mg / dl Creatina quinasa: 18 U / l; lactato deshidrogenasa: 237U / lMU: proteínas 150 mg / dl; glóbulos rojos 40,8 / campo; proteinuria: 1452 mg / 24 h C3: 36 mg / dl; C4: 15 mg / dl; anti-dsDNA: 200 UI / ml Anticoagulante lúpico, anticardiolipinas, beta 2-glicoproteína 1: negativo Biopsia renal; glomerulonefritis proliferativa lupus focal clase II (ISN / RPS) con un índice de cronicidad y actividad de 4 |
| Readmisión | HB: hemoglobina 8,90 mg / dl; hematocrito 26,3%; plaquetas 130.000 / μl Creatinina 2,77 mg / dl (eGFR CKD-EPI: 22,8 ml / min / 1,73 m2); urea 105,1 mg / dl Lactato deshidrogenasa: 527 U / l; γ-glutamil transferasa 79U / l; fosfatasa alcalina: 138 U / l; bilirrubina total: 1,23 mg / dl; directo: 0,50 mg / dl; indirecto: 0,73 mg / dl MU: proteínas 300 mg / dl; glóbulos rojos 44 / campo; proteinuria 1974mg / 24h Tomografía simple de cráneo: hipodensidad occipital derecha, sin efecto masa. No hay signos de isquemia o sangrado Electroencefalograma: actividad cerebral de fondo a 4 Hz en ritmo theta de bajo voltaje. Presencia de ondas de vértice agudas con ritmos delta posteriores con simetría interhemisférica, sin actividad epileptiforme paroxística Angiografía por resonancia magnética del cerebro en fases arterial y venosa: sin alteraciones vasculares |
Anti-dsDNA: ADN anti-bicatenario; HB: biometría hemática; MU: análisis de orina microscópico; ISN / RPS: Sociedad Internacional de Nefrología / Sociedad de Patología Renal; LES: lupus eritematoso sistémico; eGFR CKD-EPI: tasa de filtración glomerular estimada utilizando la ecuación de la Colaboración Enfermedad Renal Crónica-Epidemiología.
Discusión
Desde la primera descripción del PRES, realizada en 1996 por Hinchey et al., se ha ampliado el conocimiento de varios aspectos de esta entidad. Su nombre original de síndrome de leucoencefalopatía posterior reversible resultó inapropiado, ya que los cambios de imagen no siempre se limitan a la sustancia blanca cerebral y sus manifestaciones clínicas no siempre son reversibles9. Los primeros 15 casos reportados ocurrieron en pacientes con encefalopatía hipertensiva, eclampsia o bajo tratamiento inmunosupresor. tratamiento.10 También se ha observado como una complicación de otras entidades como sepsis, insuficiencia renal y trastornos del tejido conectivo; por lo tanto, actualmente se sabe que los factores de riesgo que causan disfunción endotelial son claves para el desarrollo de PRES.11 Se desconoce la incidencia global, pero datos de estudios retrospectivos indican que es más frecuente en individuos entre 39 y 47 años, generalmente mujeres, con comorbilidades tales como trastornos hipertensivos, renales o autoinmunes.12 En pacientes con LES, muchos autoanticuerpos se dirigen contra el endotelio; produciendo su activación, expresión de moléculas de adhesión (E-selectina, VCAM-1, ICAM-1) y exposición a citocinas proinflamatorias como IL-1β, TNFα e IL-6, provocando disrupción de la BHE y aparición de complicaciones neurológicas13. Se ha informado que en personas diagnosticadas con LES, el PRES ocurre en el contexto de actividad lúpica moderada a severa, así como asociado con insuficiencia renal e hipertensión mal controlada.14
En cuanto a las manifestaciones clínicas del PRES, se caracteriza por grados variables de encefalopatía, desde confusión hasta estupor (50-80%), convulsiones (60-75%), cefalea (50%) y alteraciones visuales que van desde visión borrosa hasta ceguera cortical (33%); siendo inusuales el déficit neurológico focal (10-15%) y el estado epiléptico (5-15%) 15. Para la valoración inicial del compromiso neurológico en estos pacientes se suele solicitar una tomografía axial computarizada (TC) craneal, que Suele ser normal o puede mostrar hipodensidades cortical-subcorticales, predominantemente en regiones cerebrales posteriores16. La RM cerebral determina el diagnóstico, mostrando edema vasogénico, generalmente en la sustancia blanca de los lóbulos occipital y parietal (territorio de la circulación cerebral posterior) , visualizadas como lesiones hiperintensas en T2 y FLAIR, bilaterales y simétricas17. La afectación preferente de la sustancia blanca se debe a su estructura de fibras mielinizadas, arteriolas y capilares que le confieren una mayor laxitud. Asimismo, los vasos de la circulación cerebral anterior, que tienen mayor inervación simpática, pueden responder adecuadamente por vasoconstricción al aumento brusco del flujo sanguíneo cerebral secundario a la hipertensión; mecanismo protector menos desarrollado en el sistema vertebrobasilar18. Con menor frecuencia, la sustancia gris y otros lóbulos pueden verse afectados. Las imágenes con secuencias de difusión permiten distinguir entre el edema vasogénico, típico del PRES, y el edema citotóxico que puede presentarse atípicamente y puede progresar a infarto19. El electroencefalograma no siempre se correlaciona con la afectación neurológica, pero puede revelar encefalopatía por la presencia de ondas focales nítidas. En pacientes con convulsiones asociadas a PRES, la principal alteración electroencefalográfica es el enlentecimiento general de las frecuencias theta / delta20. El análisis del líquido cefalorraquídeo muestra cambios inespecíficos como un ligero aumento de celularidad y proteínas, por lo que es útil cuando es conveniente para descartar una infección en el sistema nervioso central21. Además de las pruebas antes mencionadas, las que se consideran necesarias para el diagnóstico diferencial, principalmente con neurolupus, encefalopatía metabólica y parainfecciosa, encefalitis, infarto de la arteria cerebral posterior y trastornos desmielinizantes 22,23 Con base en lo anterior, en la figura 3 se muestra el algoritmo propuesto por Fugate et al. para el diagnóstico de PRES, que tiene como objetivo identificar incluso casos atípicos.15
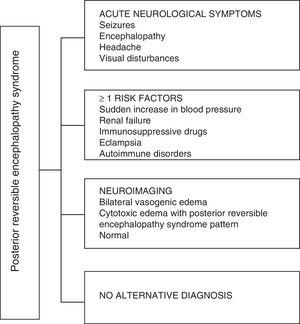
Algoritmo de diagnóstico para el síndrome de encefalopatía posterior reversible.
En nuestro paciente, dada la presentación clínica junto con los múltiples factores de riesgo, hallazgos de imagen y exclusión de otras etiologías, el diagnóstico de PRES fue concluido. Se instauró oportunamente tratamiento sintomático con medicación anticonvulsiva y anti-edema cerebral, junto con el control de los factores causales: hipertensión severa, LES con actividad severa, glomerulonefritis lúpica, fármacos inmunosupresores; ratificando el diagnóstico durante el seguimiento con la resolución de las alteraciones clínicas y de imagen. Con respecto al manejo del PRES, se debe reducir la presión arterial, se deben tratar las convulsiones y se debe controlar el desencadenante. La rápida disminución de la presión arterial podría causar isquemia cerebral, por lo que se sugiere un objetivo de una presión arterial media entre 105 y 125 mmHg, sin superar el 25% de esta reducción en la primera hora. Los fármacos de primera línea son los bloqueadores de los canales de calcio (nicardipina o nimodipina de elección, que también previene el vasoespasmo cerebral) o los betabloqueantes (por ejemplo, labetalol). El nitroprusiato de sodio o la hidralazina se pueden usar como medicamentos de segunda línea. Debe evitarse la nitroglicerina por su efecto vasodilatador, que aumentaría el edema cerebral24. El tratamiento de las crisis es similar al de otras crisis epilépticas. Las benzodiazepinas como el lorazepam o el diazepam se utilizan como terapia de primera línea. Como segunda línea, fenitoína o valproato, especialmente en el estado epiléptico, o fenobarbital. El sulfato de magnesio se puede utilizar en mujeres embarazadas. En las convulsiones refractarias, podemos administrar propofol o pentobarbital.25 En pacientes con LES deben evitarse los fármacos que podrían provocar lupus inducido por fármacos, como hidralazina, metildopa, captopril, fenitoína, valproato y carbamazepina. Existe controversia sobre el manejo de los fármacos inmunosupresores en el tratamiento del SEPR en pacientes con LES26. Tras la resolución del SEPR, las convulsiones son infrecuentes, por lo que se debe considerar la suspensión de los anticonvulsivos siempre que exista un adecuado control los factores de riesgo.Con un tratamiento oportuno y adecuado, la mayoría de los pacientes con PRES evolucionan satisfactoriamente con remisión de los síntomas y lesiones imagenológicas en pocos días o semanas, aunque se han observado complicaciones, especialmente hemorrágicas, en el 9-33% de los casos, por lo que Este caso destaca la importancia de su reconocimiento y manejo, que a menudo es un desafío.28
Conclusiones
El diagnóstico de PRES requiere una alta sospecha clínica y de imagen. El tratamiento oportuno con control de los síntomas y la causa subyacente ratifica el diagnóstico durante el seguimiento, con la resolución de las alteraciones clínicas y de imagen; de lo contrario, puede provocar secuelas neurológicas o la muerte.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.