På denna sida:
- Vad är multipel endokrin neoplasi typ 1?
- Hur vanligt är MEN1?
- Vem är mer benägna att utveckla MEN1?
- Vilka är komplikationerna med att ha MEN1?
- Vilka är symtomen på MEN1?
- Vad orsakar MEN1?
- Hur diagnostiserar läkare MEN1?
- Hur behandlar läkare MEN1?
- Hur kan genetisk rådgivning hjälpa till?
- Kliniska prövningar för MEN1
Vad är multipel endokrin neoplasi typ 1?
Multipel endokrin neoplasi typ 1 (MEN1) är en sällsynt genetisk störning som främst påverkar de endokrina körtlarna . Dessa körtlar ligger i olika delar av kroppen och kontrollerar produktionen av hormoner som styr många kroppsprocesser, inklusive tillväxt, matsmältning och sexuell funktion.
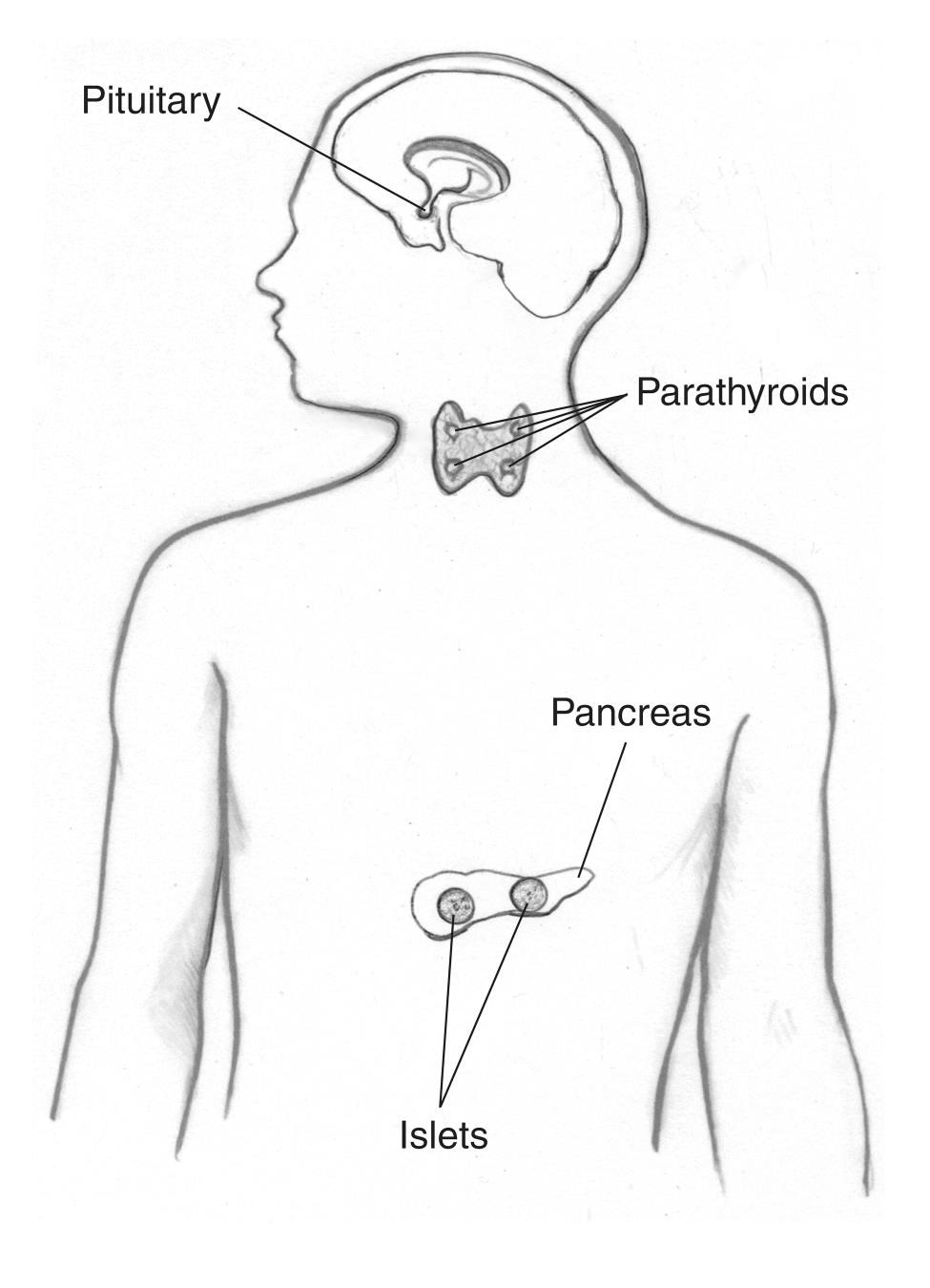
Tidigare kallad Wermers syndrom, MEN1 får tumörer att utvecklas i
- bisköldkörteln
- hypofysen
- bukspottkörteln och andra delar av mag-tarmkanalen, såsom duodenum och mage
Personer med MEN1 kan också utveckla tumörer – vanligtvis godartade (inte cancer) – i andra endokrina körtlar och kroppsvävnader, inklusive huden. Flera tumörer utvecklas ofta samtidigt i olika vävnader.
Hur vanligt är MEN1?
MEN1 är ett sällsynt, ärftligt tillstånd som förekommer hos cirka 1 av 30 000 personer.1
Vem är mer benägna att utveckla MEN1?
En familjehistoria av sjukdomen ökar din risk. Om en av dina föräldrar har genen för MEN1 har du 50 procents chans att ärva den defekta genen.
MEN1 påverkar män och kvinnor lika. Även om störningen kan drabba alla åldersgrupper, är de första symptomen vanligtvis kopplade till överaktiva bisköldkörtlar och uppträder ofta hos personer i början av 20-talet.2,3 De flesta diagnostiseras som att de har MEN1 i 40-talet, när sjukdomen har börjat påverka andra endokrina körtlar.
Vilka är komplikationerna med att ha MEN1?
MEN1 får tumörer att utvecklas i endokrina körtlar och andra delar av kroppen. Även om de flesta av dessa tumörer är cancerframkallande kan de orsaka att de drabbade körtlarna ökar i storlek och blir överaktiva och producerar för mycket hormon. I vissa fall kan en stor tumör orsaka att en körtel blir underaktiv eller inte kan producera tillräckligt med hormon.
Komplikationerna varierar beroende på
- tumörernas placering
- storleken på tumörerna
- påverkade hormon (er), om någon
- oavsett om tumörerna är cancerframkallande eller inte
Vissa tumörer fungerar inte, vilket innebär att de inte producerar hormoner. När de är små kan dessa tumörer inte orsaka komplikationer.
Bisköldkörteln
Cirka 95 procent av personerna med MEN1 utvecklar tumörer i bisköldkörtlarna efter 50 års ålder. Dessa fyra ärtkörtlar producerar parathormon, vilket hjälper till att upprätthålla rätt balans mellan kalcium och fosfor i kroppen. Med tiden kan MEN1 påverka alla fyra körtlar.
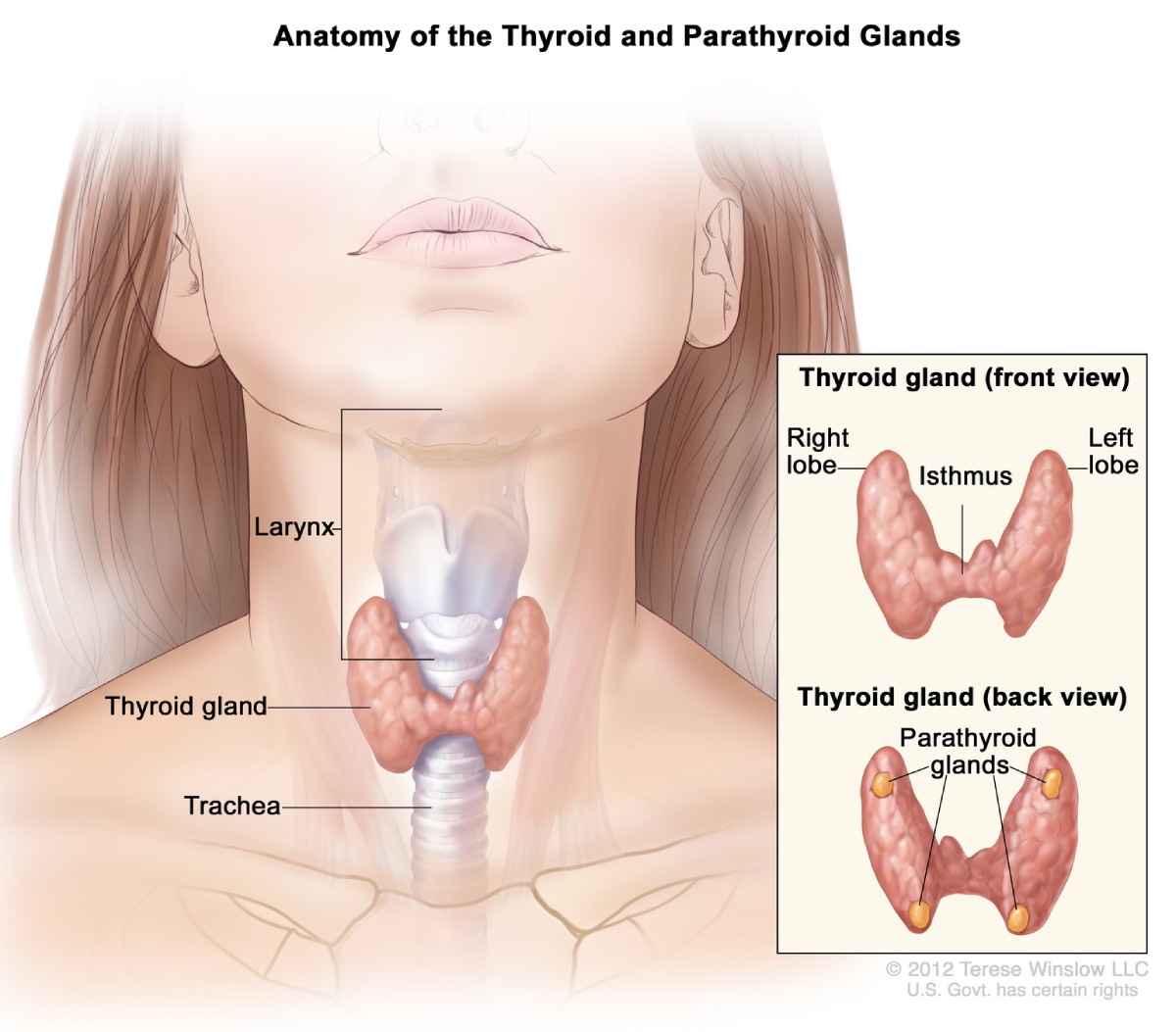
Hyperparatyreoidism. MEN1-relaterade tumörer gör att bisköldkörtlarna blir överaktiva och producerar för mycket bisköldkörtelhormon. Detta tillstånd, kallad hyperparatyreoidism, är den vanligaste komplikationen i samband med MEN1. Överdrivet bisköldkörtelhormon gör att kalciumnivåerna i blodet stiger för högt. Komplikationer kan inkludera benförlust och njursten.
Bukspottkörteln och mag-tarmkanalen
Cirka 40 procent av personerna med MEN1 utvecklar cancer i bukspottkörteln, tolvfingertarmen eller andra delar av matsmältningskanalen. 2 Många olika typer av små tumörer kan utvecklas samtidigt. Många av dessa tumörer producerar hormoner, medan andra inte producerar hormoner. Vissa tumörer kan vara cancerösa.
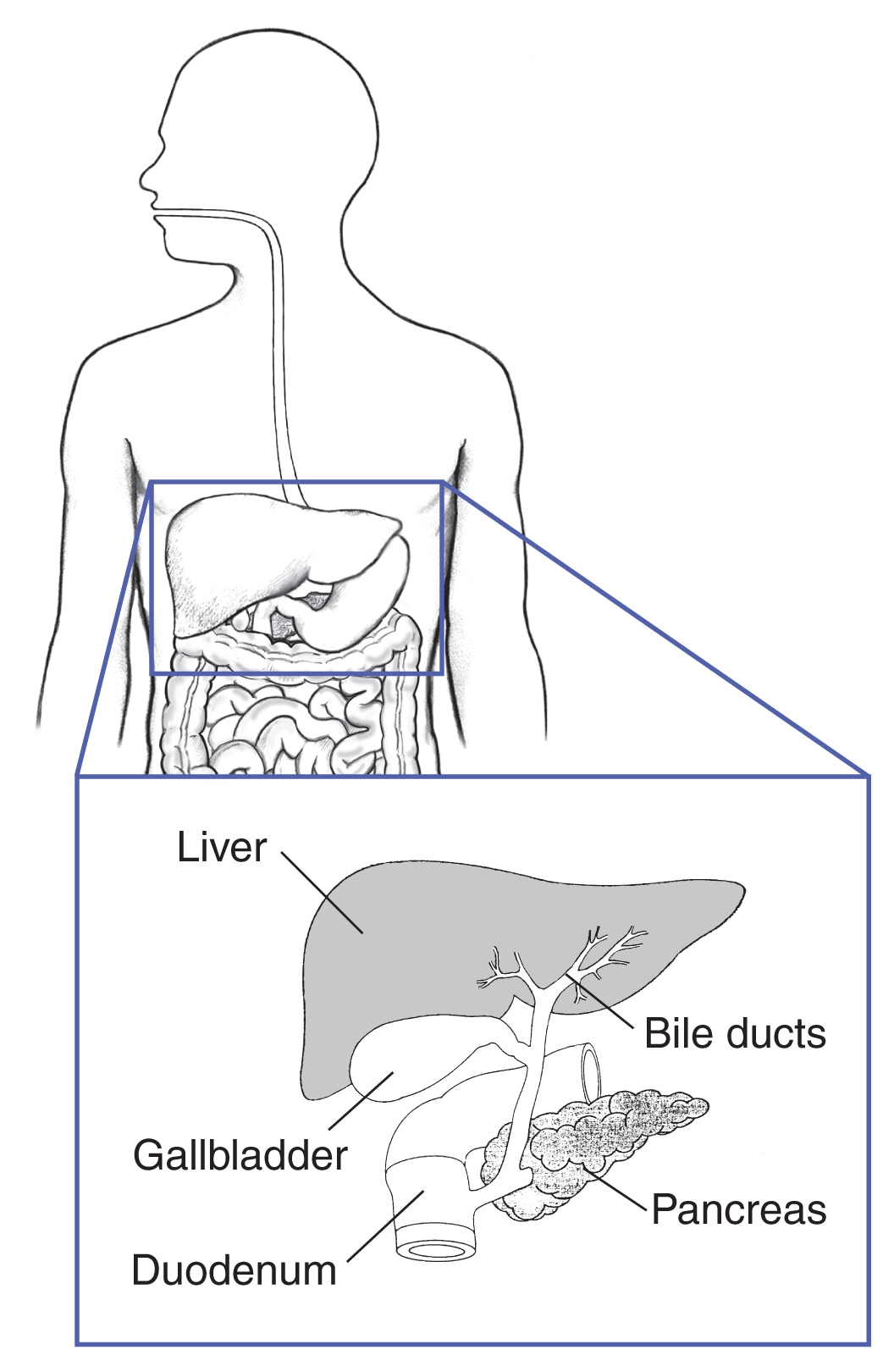
Hos personer med MEN1 är de två vanligaste tumörerna i mag-tarmkanalen
- Gastrinom . Dessa tumörer producerar hormonet gastrin, vilket får magen att frigöra syra som hjälper magen att smälta mat. För mycket gastrin kan orsaka magsår och allvarlig diarré, vilket leder till ett tillstånd som kallas Zollinger-Ellisons syndrom. Människor med MEN1 har ofta många små gastrinom – oftast i tolvfingertarmen men också i bukspottkörteln. Med tiden kan vissa av dessa tumörer bli cancerösa.
- Insulinom. Dessa tumörer bildas endast i bukspottkörteln, i celler som producerar hormonet insulin.Insulin kontrollerar nivåerna av blodsocker (blodsocker) genom att flytta glukos in i cellerna, där det kan användas för energi. Insulinom gör för mycket insulin, vilket leder till lågt blodsocker. Dessa tumörer är nästan alltid cancerframkallande och kan vanligtvis tas bort med kirurgi.
Andra, mer sällsynta bukspottskörteltumörer kan också utvecklas och orsaka andra komplikationer. Dessa tumörer inkluderar
- Glukagonomas. Dessa tumörer gör att celler i bukspottkörteln producerar för mycket av hormonet glukagon, vilket höjer blodsockret.
- VIPomas. Dessa tumörer får celler i bukspottkörteln att producera ett hormon som kallas vasoaktiv tarmpeptid (VIP), som släpper ut vatten i tarmen.
Hypofysen
Nästan 1 av 3 personer med MEN1 utvecklar tumörer i hypofysens främre del, kallad främre lob.2 Liksom andra hypofystumörer är dessa tillväxt ofta små i storlek och är nästan alltid godartade.
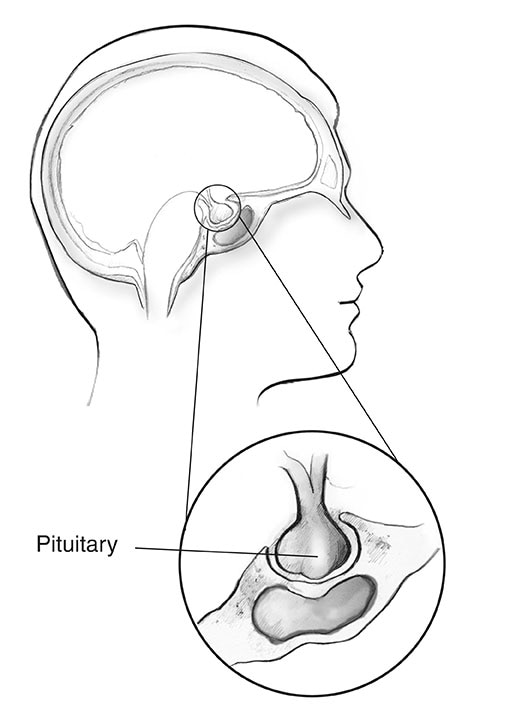
Hos personer med MEN1 är de två vanligaste hypofystumörerna
- Prolactinomas. Den vanligaste hypofystumören hos personer med MEN1, prolaktinom producerar hormonet prolaktin. Normalt signalerar detta hormon kvinnors bröst för att producera mjölk under graviditet och amning. Kvinnor med prolaktinom kan märka urladdning av mjölk från bröstet (kallas galaktorré) när de inte är gravida eller ammar. Komplikationer från att ha för mycket prolaktin i blodet kan inkludera infertilitet och benförlust.
- Tumörer som producerar tillväxthormon (GH). GH-producerande tumörer är de näst vanligaste hypofystumörerna hos personer med MEN1.4 Överskott av GH får ben och andra kroppsvävnader att växa sig större, vilket kan orsaka ett tillstånd som kallas akromegali. Relaterade hälsoproblem kan inkludera artrit, karpaltunnelsyndrom, tumörer i tjocktarmen eller ändtarmen och hjärtsjukdomar.
Vissa tumörer kan producera både prolaktin och GH. Andra, mer sällsynta hypofystumörer kan producera andra hormoner, vilket kan leda till olika symtom och komplikationer. Dessa hormoner inkluderar kortisol, som hjälper din kropp att reagera på stress och sköldkörtelhormoner som påverkar ämnesomsättningen.
Hypofystumörer som blir stora i storlek kan orsaka andra problem, vilket gör det svårt för hypofysen att fungera ordentligt. Dessa tumörer kan förhindra att hypofysen producerar tillräckligt med hormoner, vilket leder till ett tillstånd som kallas hypopituitarism. Tumörerna kan också pressa mot närliggande hjärnvävnader och orsaka huvudvärk och / eller synproblem.
Andra tumörer
MEN1 kan också orsaka tumörer i andra delar av kroppen. Exempel inkluderar
- tumörer i andra endokrina körtlar, såsom binjurarna
- carcinoida tumörer – långsamt växande tumörer som oftast finns i mage, tymus och lungor
- hudtumörer och tumörer under huden, oftast angiofibrom, lipom (godartade tumörer gjorda av fettceller) och kollagenom (tumörer som involverar ett protein i huden som kallas kollagen)
- meningiom och ependymom —Tumörer av celler som sträcker hjärnan och ryggmärgen
Komplikationer kan variera beroende på typ, storlek och plats för tumören.
Vilka är symtomen av MEN1?
MEN1-symtom kan skilja sig från person till person, även bland familjemedlemmar som har sjukdomen. Åldern vid vilken tecken eller symtom uppträder kan också variera.
Bisköldkörteltumörer
Eftersom MEN1 nästan alltid påverkar bisköldkörteln, är de vanligaste tidiga symptomen relaterade till överskott av bisköldkörtelhormon. Dessa symtom är ofta milda och kanske inte märks på flera år. De inkluderar
- njurstenar
- muskelsvaghet
- trötthet
- ökad törst och urinering
- depression
- värk i ben och leder
- matsmältningsproblem och förstoppning

Andra tumörer
Tumörer i andra endokrina körtlar kan orsaka andra symtom, såsom
- magsår
- surt återflöde
- buksmärta
- frekvent diarré
- lågt blodsocker
- förstorade och svullna händer och fötter
Vad orsakar MEN1?
MEN1 är en ärftlig sjukdom som oftast orsakas av en mutation i MEN1-genen. Genen ger instruktioner för att producera ett protein som kallas menin, känt för att spela en roll för att förhindra att celler växer och delar sig för snabbt.
MEN1 är en autosomal dominerande störning. Detta innebär att endast en förälder behöver ha den defekta genen för att överföra störningen till ett barn. Om en förälder har MEN1-genen har varje barn 1 till 2 (50%) chans att få sjukdomen.I ungefär 1 av 10 fall ärvs inte mutationen från någon av föräldrarna utan utvecklas på egen hand. Detta är en naturlig, slumpmässig process som kan förekomma hos vem som helst.4
Varje person i familjen som har MEN1-syndrom delar samma mutation. Genom att studera olika familjer med MEN1 har forskare identifierat hundratals olika mutationer av MEN1-genen som kan orsaka störningen. Om du har någon av dessa mutationer betraktas du som bärare av MEN1, även om du inte har några symtom.
Att veta om du är bärare är viktigt eftersom det är mycket troligt att du utvecklar även utan symtom några MEN1-relaterade tumörer under din livstid. Du kan redan ha utvecklat tumörer som inte har upptäckts om du inte har gjort en grundlig bedömning och du kan fortfarande ge sjukdomen vidare till ett barn.
Om det finns en känd MEN1-mutation i din familj och genetisk testning visar att du inte bär det, då har du inte MEN1-syndrom. Det är osannolikt att du utvecklar MEN1-relaterade tumörer under din livstid och du kommer inte att överföra sjukdomen till några barn.
Hur diagnostiserar läkare MEN1?
Din läkare kommer att diagnostisera dig som att ha MEN1 om du uppfyller ett av dessa tre kriterier5
- två eller flera MEN1-relaterade tumörer (tumörer i bisköldkörteln, hypofysen och bukspottkörteln eller någon annan del av mag-tarmkanalen)
- en MEN1-relaterad tumör och en första grads släkting (en förälder, bror eller syster eller barn) som kliniskt har diagnostiserats som att ha MEN1
- en MEN1-mutation, även om du har inga tecken eller symtom på MEN1
Genetisk testning för MEN1-mutation
Genetisk testning hjälper dig att ta reda på om du har en genmutation som är känd för att orsaka MEN1. Test rekommenderas för 5
- personer som har två eller flera MEN1-relaterade endokrina tumörer eller andra tecken eller symtom på MEN1
- alla första grads släktingar till en person som har MEN1-genmutation
En tidig diagnos hjälper dig att övervaka dina symtom och ta itu med problem innan de blir allvarliga. Genetiska tester för en känd familjemutation kan vara lämpliga från och med 5 års ålder, eftersom barn med MEN1 i sällsynta fall kan utveckla tumörer i hypofysen eller bisköldkörteln. Den typiska startåldern för MEN1-syndrom är i tonåren eller 20-talet, men de första tumörerna hos någon med MEN1 kan utvecklas tidigare eller senare. Symtom och typer av tumörer kan skilja sig åt bland medlemmar i samma familj.
Genetisk testning utförs oftast på ett blodprov. Vissa laboratorier kan också använda saliv eller en svabb på insidan av kinden för att utföra denna testning.

I upp till 1 av 4 fall kanske testet inte hittar en mutation även om du kanske visar tecken på störningen.2 I dessa fall kan orsaken vara en okänd MEN1-mutation eller en mutation i en annan gen. Om ditt test inte hittar en MEN1-relaterad mutation kan din läkare beställa andra tester för att ta reda på om dina symtom beror på en annan orsak.
Hur behandlar läkare MEN1?
Även om MEN1 inte kan botas, lever de flesta med sjukdomen långa och produktiva liv. Din läkare kommer att övervaka din hälsa och tillhandahålla behandling efter behov.
Hantera symtom och övervaka tumörer
Din läkare kommer att övervaka dina symtom och screena för tecken på MEN1-relaterade tumörer regelbundet grund. Vanligt använda screeningtester inkluderar
- Blodprover. Dessa tester hjälper din läkare att övervaka nivåer av hormoner och andra ämnen kopplade till MEN1-relaterade tumörer. Exempel inkluderar kalcium och paratyroidhormon, prolaktin, gastrin och markörer för vissa tumörer.
- Bildtest. Din läkare kan beställa avbildningstester för att övervaka storlek och tillväxt av befintliga tumörer och för att upptäcka nya, inklusive tumörer som inte frisätter hormoner eller orsakar symtom. Dessa tester inkluderar magnetisk resonanstomografi (MRI), datortomografi (CT) och ultraljud. Andra tester som letar efter onormala hormonreceptorer – proteiner som fäster vid vissa hormoner – på ytan av tumörer kan hjälpa till att upptäcka tumörer som kanske inte syns vid en MR- eller CT-skanning.
Baserat på dina symtom och testresultat kan din läkare ordinera olika läkemedel för att hantera sjukdomens framsteg.
Behandling av tumörer
Om tumörerna är små och inte orsakar symtom kan ingen behandling vara behövs. Läkare kommer att följa dessa tumörer med blod- och bildtest.
Behandlingen varierar beroende på plats och typ av tumör. Till exempel
- Paratyroidtumörer behandlas oftast med kirurgi för att avlägsna de drabbade körtlarna. Om kirurgi inte är möjligt kan din läkare ordinera läkemedel för att kontrollera kalciumnivåerna.
- Tumörer i bukspottkörteln och matsmältningsorganet behandlas ofta med läkemedel för att kontrollera symtom som för mycket magsyra.Andra behandlingsalternativ inkluderar kirurgi för att avlägsna tumören, frysa eller bränna tumörer som har spridit sig till levern utan att ta bort dem och, mer sällan, systemisk kemoterapi – behandling med cancerläkemedel som reser genom blodet till celler över hela kroppen .
- Hypofystumörer behandlas oftast med läkemedel och / eller kirurgi. Strålbehandling kan också användas, men mer sällan.
Behandling av flera tumörer. Människor med MEN1 utvecklar ofta många tumörer samtidigt. Som ett resultat är behandlingen mer komplicerad än bland personer som har en enda tumör eller mycket få tumörer. Ibland kan MEN1-relaterade tumörer vara större, mer aggressiva och motståndskraftiga mot behandling än andra tumörer.2
Kirurgi för att ta bort tumörer. Kirurgi lyckas ofta med att ta bort MEN1-relaterade tumörer och bota relaterade symtom. Men i vissa fall kan tumörerna växa tillbaka eller sprida sig till lymfkörtlar, levern eller, mer sällan, benen. Din läkare kan ordinera läkemedel för att minska tumörstorleken och behandla relaterade problem.
Efterkirurgisk behandling. Om en operation tar bort en hel endokrin körtel – eller mer än tre bisköldkörtlar – kan din läkare ordinera läkemedel för att ersätta de hormoner som din kropp inte längre gör. Du kan också behöva ta andra läkemedel och kosttillskott, såsom kalcium och D-vitamin, för att åtgärda dessa brister.
Hur kan genetisk rådgivning hjälpa till?
Genetisk rådgivning är en informationskälla och stöd till familjer som drabbas av eller riskerar en genetisk störning. Till exempel kan genetiska rådgivare hjälpa dig och din familj
- förstå hur genetisk testning görs
- väga de medicinska, sociala, ekonomiska och etiska beslut som är involverade i att bli testade
- diskutera tillgängliga alternativ om hur man hanterar sjukdomen
- fatta välgrundade beslut om huruvida man ska få barn och diskutera alternativ för att testa ett barn, ett foster eller ett embryo för en känd familjemutation i MEN1
- ta reda på vilka familjemedlemmar som är i riskzonen och kan dra nytta av genetisk testning av en känd familjär MEN1-mutation
Genetiska rådgivare kan också hänvisa dig till ett intervall av supporttjänster, inklusive utbildningskällor, förespråks- och supportgrupper, annan vårdpersonal och lokala eller statliga tjänster.
Du kan hitta genetiska rådgivare nära dig med sökverktyget Hitta en genetisk rådgivare från National Society for Genetiska rådgivare. När du använder verktyget, under ”Typer av specialisering”, välj ”Cancer”.
Kliniska prövningar för MEN1
NIDDK genomför och stöder kliniska prövningar i många sjukdomar och tillstånd, inklusive endokrin sjukdomar. Prövningarna försöker hitta nya sätt att förebygga, upptäcka eller behandla sjukdomar och förbättra livskvaliteten.
Vad är kliniska prövningar för MEN1?
Kliniska prövningar – och andra typer av kliniska prövningar studier – är en del av medicinsk forskning och involverar människor som du. När du frivilligt deltar i en klinisk studie hjälper du läkare och forskare att lära sig mer om sjukdomar och förbättra hälsovården för människor i framtiden.
Forskare studerar många aspekter av MEN1, inklusive nya behandlingar för detta tillstånd.
Ta reda på om kliniska studier passar dig.
Vilka kliniska studier för MEN1 letar efter deltagare?
Du kan visa en filtrerad lista över kliniska studier på MEN1 som är öppna och rekryterar på
www.ClinicalTrials.gov. Du kan utöka eller begränsa listan så att den inkluderar kliniska studier från industri, universitet och individer. National Institutes of Health granskar dock inte dessa studier och kan inte säkerställa att de är säkra. Prata alltid med din vårdgivare innan du deltar i en klinisk studie.