På denne side:
- Hvad er multipel endokrin neoplasi type 1?
- Hvor almindelig er MEN1?
- Hvem er mere tilbøjelige til at udvikle MEN1?
- Hvad er komplikationerne ved at have MEN1?
- Hvad er symptomerne på MEN1?
- Hvad forårsager MEN1?
- Hvordan diagnosticerer læger MEN1?
- Hvordan behandler læger MEN1?
- Hvordan kan genetisk rådgivning hjælpe?
- Kliniske forsøg med MEN1
Hvad er multipel endokrin neoplasi type 1?
Multipel endokrin neoplasi type 1 (MEN1) er en sjælden genetisk lidelse, der primært påvirker de endokrine kirtler . Disse kirtler er placeret i forskellige dele af kroppen og styrer produktionen af hormoner, der styrer mange kropsprocesser, herunder vækst, fordøjelse og seksuel funktion.
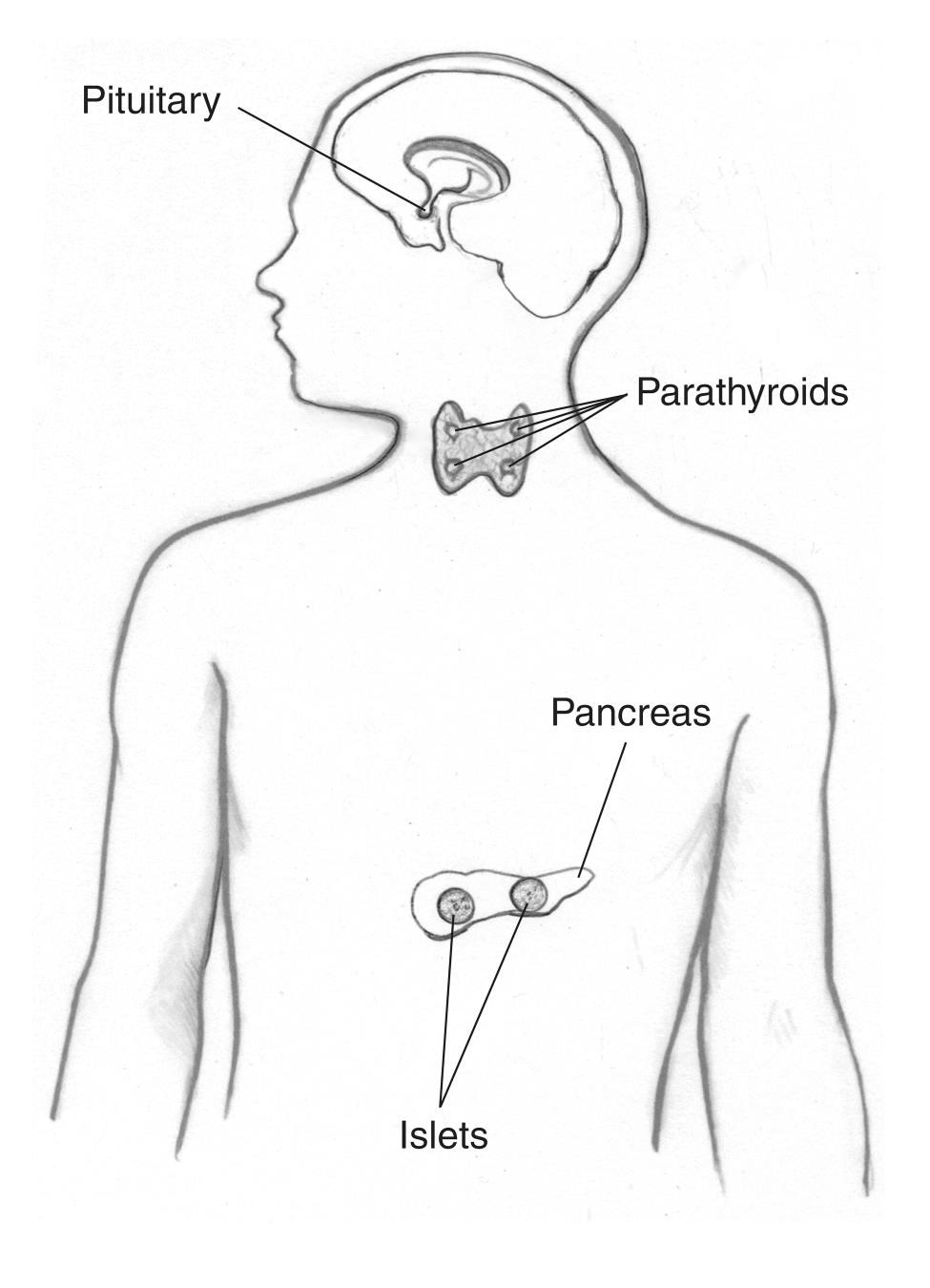
Tidligere kaldet Wermers syndrom, MEN1 får tumorer til at udvikle sig i
- parathyroidea kirtler
- hypofysen
- bugspytkirtlen og andre dele af fordøjelseskanalen, såsom tolvfingertarmen og mave
Mennesker med MEN1 kan også udvikle tumorer – normalt godartede (ikke kræft) – i andre endokrine kirtler og kropsvæv, herunder huden. Flere tumorer udvikler sig ofte på samme tid i forskellige væv.
Hvor almindelig er MEN1?
MEN1 er en sjælden, arvelig tilstand, der forekommer hos ca. 1 ud af 30.000 mennesker.1
Hvem er mere tilbøjelige til at udvikle MEN1?
En familiehistorie af lidelsen øger din risiko. Hvis en af dine forældre har genet for MEN1, har du 50 procent chance for at arve det defekte gen.
MEN1 påvirker mænd og kvinder lige meget. Selvom forstyrrelsen kan påvirke alle aldersgrupper, er de første symptomer typisk forbundet med overaktive biskjoldbruskkirtler og forekommer ofte hos mennesker i de tidlige 20ere. andre endokrine kirtler.
Hvad er komplikationerne ved at have MEN1?
MEN1 får tumorer til at udvikle sig i endokrine kirtler og andre dele af kroppen. Selvom de fleste af disse tumorer ikke er kræftfremkaldende, kan de få de berørte kirtler til at stige i størrelse og blive overaktive og producere for meget hormon. I nogle tilfælde kan en stor tumor få en kirtel til at blive underaktiv eller ude af stand til at producere nok hormon.
Komplikationer varierer afhængigt af
- placeringen af tumorer
- størrelsen af de tumorer, der er påvirket
- påvirket hormon (er), hvis nogen
- uanset om tumorer er kræftfremkaldende eller ikke
Nogle tumorer fungerer ikke, hvilket betyder, at de ikke producerer hormoner. Når disse tumorer er små, forårsager de muligvis ingen komplikationer.
Biskjoldbruskkirtler
Cirka 95 procent af mennesker med MEN1 udvikler svulster i biskjoldbruskkirtlerne i alderen 50,2. Disse fire kirtler i ørestørrelse producerer parathyroideahormon, som hjælper med at opretholde den rette balance mellem calcium og fosfor i din krop. Over tid kan MEN1 påvirke alle fire kirtler.
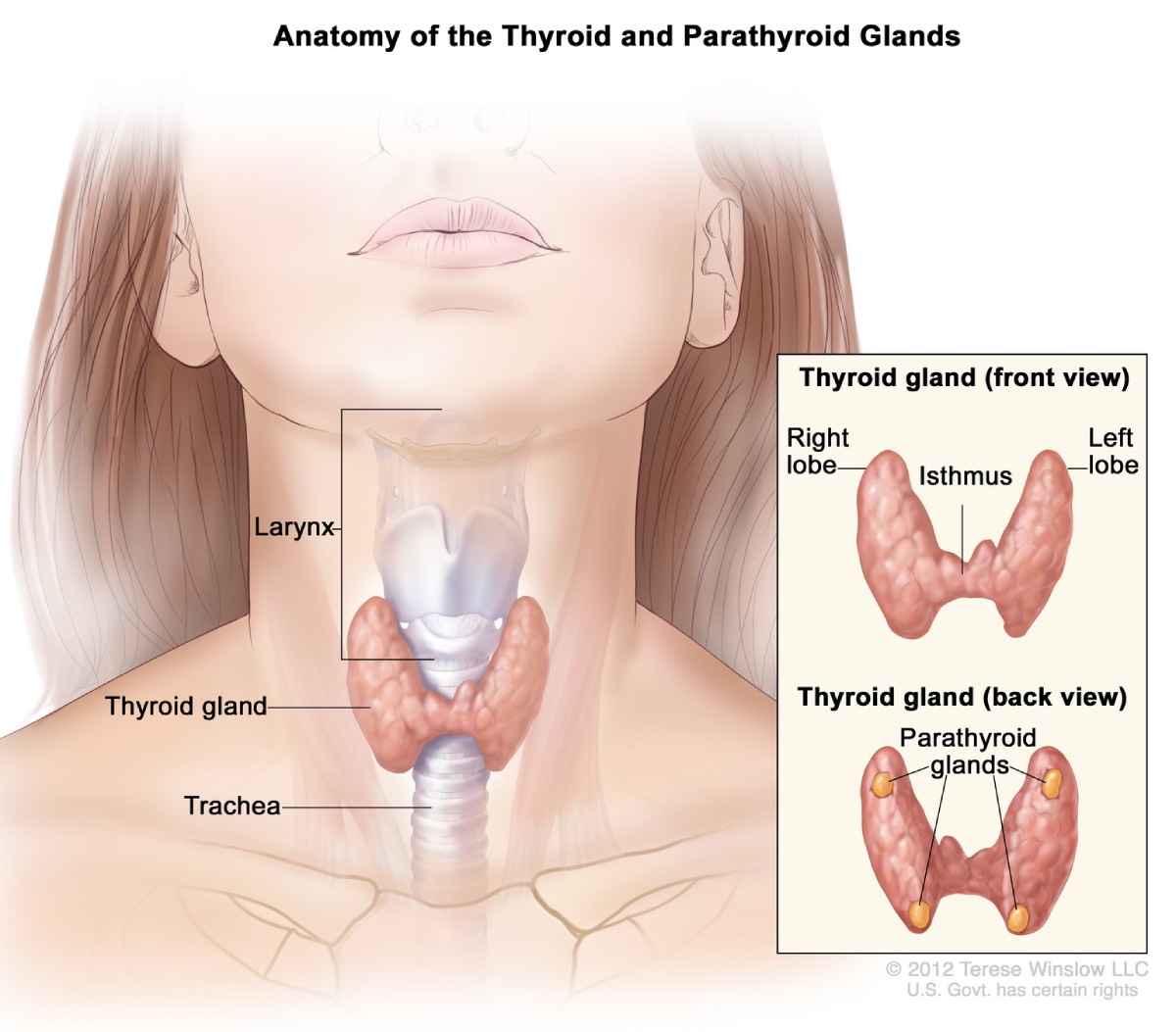
Hyperparathyroidisme. MEN1-relaterede tumorer får biskjoldbruskkirtlen til at blive overaktiv og producerer for meget biskjoldbruskkirtelhormon. Denne tilstand, kaldet hyperparatyreoidisme, er den mest almindelige komplikation forbundet med MEN1. Overskydende parathyroideahormon får calciumniveauerne i dit blod til at stige for højt. Komplikationer kan omfatte knogletab og nyresten.
Pankreas og fordøjelseskanalen
Ca. 40 procent af mennesker med MEN1 udvikler kræft i bugspytkirtlen, tolvfingertarmen eller andre dele af fordøjelseskanalen. 2 Mange forskellige typer små tumorer kan udvikle sig på samme tid. Mange af disse tumorer producerer hormoner, mens andre ikke producerer hormoner. Nogle tumorer kan være kræftfremkaldende.
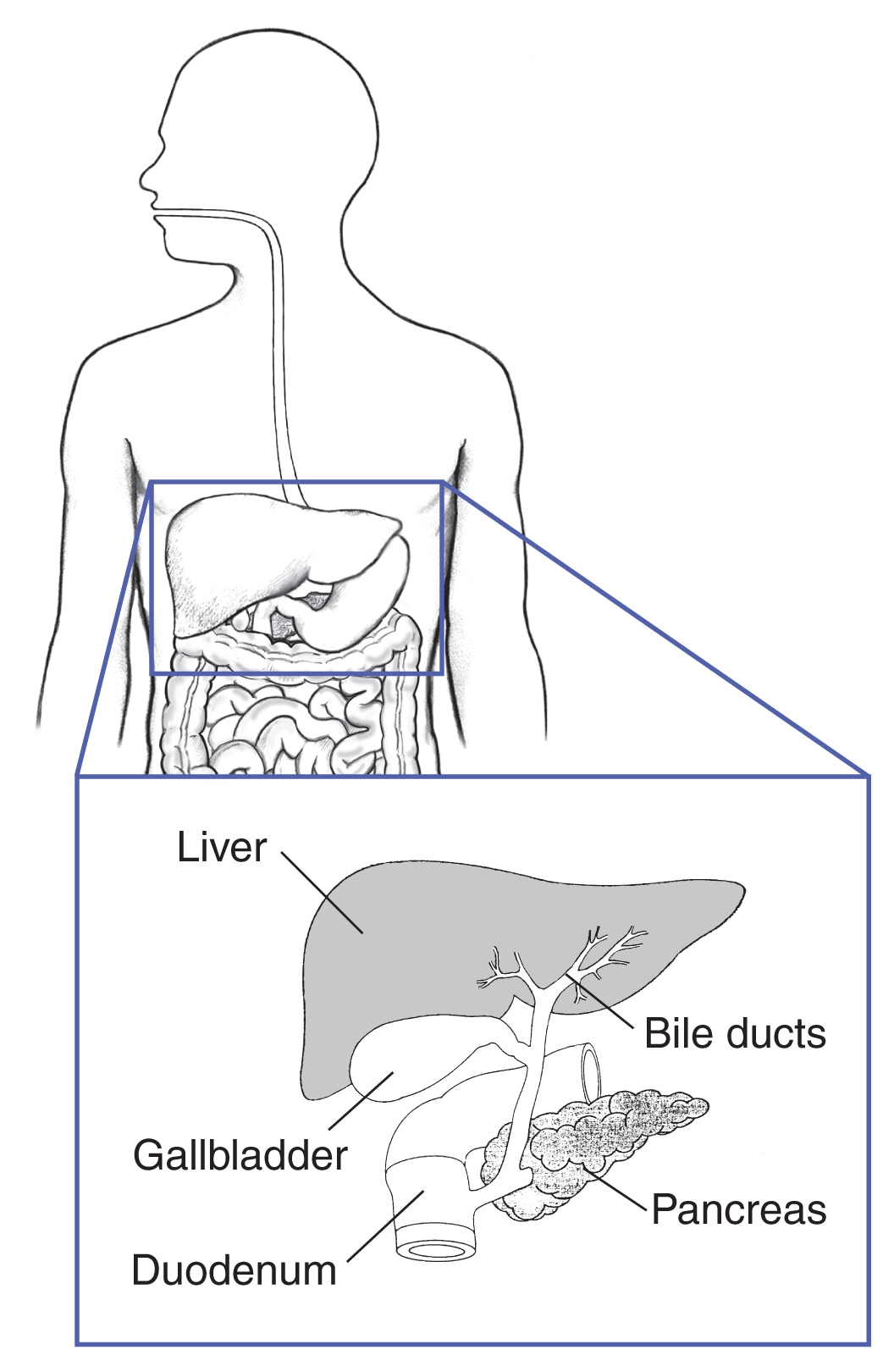
Hos mennesker med MEN1 er de to mest almindelige tumorer i fordøjelseskanalen
- Gastrinomer . Disse tumorer producerer hormonet gastrin, som får maven til at frigive syre, der hjælper maven med at fordøje mad. For meget gastrin kan forårsage mavesår og alvorlig diarré, hvilket fører til en tilstand kaldet Zollinger-Ellison syndrom. Mennesker med MEN1 har ofte mange små gastrinomer – oftest i tolvfingertarmen, men også i bugspytkirtlen. Over tid kan nogle af disse tumorer blive kræftfremkaldende.
- Insulinomer. Disse tumorer dannes kun i bugspytkirtlen, i celler, der producerer hormonet insulin.Insulin styrer niveauet af blodsukker (blodsukker) ved at flytte glukose ind i cellerne, hvor det kan bruges til energi. Insulinomer producerer for meget insulin, hvilket fører til lavt blodsukker. Disse tumorer er næsten altid ikke-kræftfremkaldende og kan normalt fjernes ved kirurgi.
Andre, mere sjældne bugspytkirteltumorer kan også udvikle sig og forårsage andre komplikationer. Disse tumorer inkluderer
- Glucagonomas. Disse tumorer får celler i bugspytkirtlen til at producere for meget af hormonet glukagon, hvilket hæver blodsukkeret.
- VIPomas. Disse tumorer får celler i bugspytkirtlen til at producere et hormon kaldet vasoaktivt tarmpeptid (VIP), som frigiver vand i tarmen.
Hypofysen
Næsten 1 ud af 3 mennesker med MEN1 udvikler tumorer i den forreste del af hypofysen, kaldet den forreste lap.2 Som andre hypofysetumorer er disse vækster ofte små i størrelse og er næsten altid godartede.
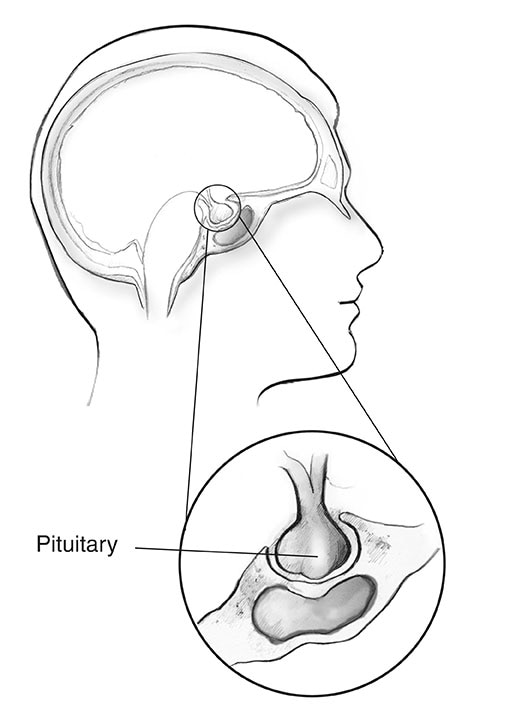
Hos mennesker med MEN1 er de to mest almindelige hypofysetumorer
- Prolactinomas. Den mest almindelige hypofysetumor hos mennesker med MEN1, prolactinomer producerer hormonet prolactin. Normalt signaliserer dette hormon kvindebrystene om at producere mælk under graviditet og amning. Kvinder med prolaktinom kan bemærke mælkeudslip fra deres bryst (e) (kaldet galactorrhea), når de ikke er gravide eller ammer. Komplikationer ved at have for meget prolactin i blodet kan omfatte infertilitet og knogletab.
- Tumorer, der producerer væksthormon (GH). GH-producerende tumorer er de næststørste hypofysetumorer hos mennesker med MEN1.4 Overskydende GH får knogler og andet kropsvæv til at vokse sig større, hvilket kan forårsage en tilstand kaldet akromegali. Relaterede sundhedsproblemer kan omfatte gigt, karpaltunnelsyndrom, tumorer i tyktarmen eller endetarmen og hjertesygdomme.
Nogle tumorer kan producere både prolactin og GH. Andre, mere sjældne hypofysetumorer kan producere andre hormoner, hvilket kan føre til forskellige symptomer og komplikationer. Disse hormoner inkluderer kortisol, som hjælper din krop med at reagere på stress og skjoldbruskkirtelhormoner, der påvirker stofskiftet.
Hypofysetumorer, der bliver store i størrelse, kan forårsage andre problemer, hvilket gør det vanskeligt for hypofysen at arbejde korrekt. Disse tumorer kan forhindre hypofysen i at fremstille nok hormoner, hvilket fører til en tilstand kaldet hypopituitarisme. Tumorerne kan også presse mod nærliggende hjernevæv og forårsage hovedpine og / eller synsproblemer.
Andre tumorer
MEN1 kan også forårsage tumorer i andre dele af kroppen. Eksempler inkluderer
- tumorer i andre endokrine kirtler, såsom binyrerne
- carcinoide tumorer – langsomt voksende tumorer, der oftest findes i mave, thymus og lunger
- hudtumorer og tumorer under huden, oftest angiofibromer, lipomer (godartede tumorer lavet af fedtceller) og collagenomer (tumorer, der involverer et protein i huden kaldet kollagen)
- meningiomas og ependymomas —Tumorer af celler, der strækker hjernen og rygmarven
Komplikationer kan variere afhængigt af tumorens type, størrelse og placering.
Hvad er symptomerne af MEN1?
MEN1 symptomer kan variere fra person til person, selv blandt familiemedlemmer, der har lidelsen. Den alder, hvor tegn eller symptomer optræder, kan også variere.
Parathyroidea tumorer
Da MEN1 næsten altid påvirker parathyroidea kirtler, er de mest almindelige tidlige symptomer relateret til overskydende parathyroideahormon. Disse symptomer er ofte milde og bemærkes muligvis ikke i årevis. De inkluderer
- nyresten
- muskelsvaghed
- træthed
- øget tørst og vandladning
- depression
- smerter i knogler og led
- fordøjelsesproblemer og forstoppelse

Andre tumorer
Tumorer placeret i andre endokrine kirtler kan forårsage andre symptomer, såsom
- mavesår
- acid reflux
- mavesmerter
- hyppig diarré
- lavt blodsukker
- forstørrede og hævede hænder og fødder
Hvad forårsager MEN1?
MEN1 er en arvelig lidelse, der oftest er forårsaget af en mutation i MEN1-genet. Genet giver instruktioner til produktion af et protein kaldet menin, der vides at spille en rolle i at forhindre celler i at vokse og dele sig for hurtigt.
MEN1 er en autosomal dominerende lidelse. Dette betyder, at kun den ene forælder skal have det defekte gen for at overføre lidelsen til et barn. Hvis en forælder har MEN1-genet, har hvert barn en 1 ud af 2 (50%) chance for at få lidelsen.I ca. 1 ud af 10 tilfælde arves mutationen ikke fra nogen af forældrene, men udvikler sig alene. Dette er en naturlig, tilfældig proces, der kan forekomme hos enhver.4
Hver person i familien, der har MEN1-syndrom, deler den samme mutation. Ved at studere forskellige familier med MEN1 har forskere identificeret hundreder af forskellige mutationer af MEN1-genet, der kan forårsage lidelsen. Hvis du har nogen af disse mutationer, betragtes du som bærer af MEN1, selvom du ikke har nogen symptomer.
At vide, om du er bærer, er vigtig, fordi du selv uden symptomer er meget tilbøjelig til at udvikle nogle MEN1-relaterede tumorer i din levetid. Du har muligvis allerede udviklet tumorer, der ikke er opdaget, hvis du ikke har haft en grundig vurdering, og du kan stadig videregive lidelsen til et barn.
Hvis der er en kendt MEN1-mutation i din familie og genetisk test viser, at du ikke bærer det, så har du ikke MEN1-syndrom. Det er usandsynligt, at du udvikler MEN1-relaterede tumorer i din levetid, og du vil ikke overføre sygdommen til nogen børn.
Hvordan diagnosticerer læger MEN1?
Din læge vil diagnosticere dig som at have MEN1, hvis du opfylder et af disse tre kriterier5
- to eller flere MEN1-relaterede tumorer (tumorer i biskjoldbruskkirtler, hypofysen og bugspytkirtlen eller anden del af fordøjelseskanalen)
- en MEN1-relateret tumor og en førstegrads slægtning (en forælder, bror eller søster eller barn), der er klinisk diagnosticeret som at have MEN1
- en MEN1-mutation, selvom du har ingen tegn eller symptomer på MEN1
Genetisk test for MEN1-mutation
Genetisk test hjælper dig med at finde ud af, om du har en genmutation, der vides at forårsage MEN1. Test anbefales til 5
- personer, der har to eller flere MEN1-relaterede endokrine tumorer eller andre tegn eller symptomer på MEN1
- alle førstegrads slægtninge til en person, der har MEN1-genmutation
En tidlig diagnose hjælper dig med at overvåge dine symptomer og løse problemer, før de bliver alvorlige. Genetisk testning af en kendt familiemutation kan være passende startende så tidligt som 5 år, fordi børn med MEN1 i sjældne tilfælde kan udvikle tumorer i hypofysen eller parathyroidea. Den typiske debutalder for MEN1-syndrom er i teenagere eller 20ere, men de første tumorer hos en person med MEN1 kan udvikle sig tidligere eller senere. Symptomerne og typerne af tumorer kan variere selv blandt medlemmer af samme familie.
Genetisk test udføres oftest på en blodprøve. Nogle laboratorier kan også bruge spyt eller en vatpind på indersiden af kinden til at udføre denne test.

I op til 1 ud af 4 tilfælde finder testen muligvis ikke en mutation, selvom du måske viser tegn på forstyrrelsen.2 I disse tilfælde kan årsagen være en ukendt MEN1-mutation eller en mutation i et andet gen. Hvis din test ikke finder en MEN1-relateret mutation, kan din læge muligvis bestille andre tests for at finde ud af, om dine symptomer skyldes en anden årsag.
Hvordan behandler læger MEN1?
Selvom MEN1 ikke kan helbredes, lever de fleste mennesker med lidelsen lange og produktive liv. Din læge vil overvåge dit helbred og yde behandling efter behov.
Håndtering af symptomer og monitorering af tumorer
Din læge overvåger regelmæssigt dine symptomer og screener for tegn på MEN1-relaterede tumorer basis. Almindeligt anvendte screeningstest inkluderer
- Blodprøver. Disse tests hjælper din læge med at overvåge niveauer af hormoner og andre stoffer forbundet med MEN1-relaterede tumorer. Eksempler inkluderer calcium og parathyreoideahormon, prolactin, gastrin og markører for visse tumorer.
- Imaging tests. Din læge kan bestille billeddannelsestest for at overvåge størrelsen og væksten af eksisterende tumorer og opdage nye, herunder tumorer, der ikke frigiver hormoner eller forårsager symptomer. Disse tests inkluderer magnetisk resonansbilleddannelse (MRI), computertomografi (CT) scanning og ultralyd. Andre tests, der ser efter unormale hormonreceptorer – proteiner, der binder sig til visse hormoner – på overfladen af tumorer kan hjælpe med at opdage tumorer, der muligvis ikke er synlige ved en MR- eller CT-scanning.
Baseret på dine symptomer og testresultater, kan din læge ordinere forskellige lægemidler til at styre sygdommens fremskridt.
Behandling af tumorer
Hvis tumorer er små og ikke forårsager symptomer, kan der ikke være nogen behandling havde brug for. Læger vil følge disse tumorer med blod- og billeddannelsestest.
Behandlingen varierer afhængigt af placeringen og typen af tumor. For eksempel
- Parathyroidea tumorer behandles oftest med kirurgi for at fjerne de berørte kirtler. Hvis kirurgi ikke er mulig, kan din læge ordinere medicin til at kontrollere calciumniveauer.
- Tumorer i bugspytkirtlen og fordøjelseskanalen behandles ofte med medicin for at kontrollere symptomer såsom for meget mavesyre.Andre behandlingsmuligheder inkluderer kirurgi for at fjerne tumor (erne), frysning eller brændende tumorer, der har spredt sig til leveren uden at fjerne dem, og mere sjældent systemisk kemoterapi – behandling med kræftlægemidler, der bevæger sig gennem blodet til celler overalt i din krop .
- Hypofysetumorer behandles oftest med medicin og / eller kirurgi. Strålebehandling kan også anvendes, men mere sjældent.
Behandling af flere tumorer. Mennesker med MEN1 udvikler ofte mange tumorer på samme tid. Som et resultat er behandlingen mere kompliceret end blandt mennesker, der har en enkelt tumor eller meget få tumorer. Nogle gange kan MEN1-relaterede tumorer være større, mere aggressive og resistente over for behandling end andre tumorer.2
Kirurgi for at fjerne tumorer. Kirurgi lykkes ofte med at fjerne MEN1-relaterede tumorer og kurere relaterede symptomer. Men i nogle tilfælde kan tumorer vokse tilbage eller sprede sig til lymfeknuder, leveren eller mere sjældent knoglerne. Din læge kan ordinere medicin for at reducere størrelsen på tumoren og behandle relaterede problemer.
Behandling efter kirurgi. Hvis en operation fjerner en hel endokrin kirtel – eller mere end tre biskjoldbruskkirtler – kan din læge ordinere medicin til erstatning for de hormoner, som din krop ikke længere fremstiller. Det kan også være nødvendigt at tage andre lægemidler og kosttilskud, såsom calcium og D-vitamin, for at afhjælpe disse mangler.
Hvordan kan genetisk rådgivning hjælpe?
Genetisk rådgivning er en kilde til information og støtte til familier, der er ramt af eller er i fare for en genetisk lidelse. For eksempel kan genetiske rådgivere hjælpe dig og din familie
- forstå, hvordan genetisk test udføres
- afveje de medicinske, sociale, økonomiske og etiske beslutninger, der er involveret i at blive testet
- diskutere tilgængelige muligheder for, hvordan man styrer sygdommen
- træffe informerede beslutninger om, hvorvidt man skal få børn, og diskutere muligheder for at teste et barn, et foster eller et foster for en kendt familiemutation i MEN1
- find ud af, hvilke familiemedlemmer der er i fare og kan drage fordel af genetisk testning af en kendt familiær MEN1-mutation
Genetiske rådgivere kan også henvise dig til en række af supporttjenester, herunder uddannelseskilder, advokat- og supportgrupper, andet sundhedspersonale og lokale eller statslige tjenester.
Du kan finde genetiske rådgivere i nærheden af dig ved hjælp af søgeværktøjet Find en genetisk rådgiver fra National Society for Genetiske rådgivere. Når du bruger værktøjet, vælges “Kræft” under “Typer af specialisering”.
Kliniske forsøg med MEN1
NIDDK udfører og understøtter kliniske forsøg i mange sygdomme og tilstande, herunder endokrin sygdomme. Forsøgene søger at finde nye måder til at forebygge, opdage eller behandle sygdomme og forbedre livskvaliteten.
Hvad er kliniske forsøg med MEN1?
Kliniske forsøg – og andre typer kliniske forsøg studier – er en del af medicinsk forskning og involverer mennesker som dig. Når du melder dig frivilligt til at deltage i en klinisk undersøgelse, hjælper du læger og forskere med at lære mere om sygdom og forbedre sundhedspleje for mennesker i fremtiden.
Forskere studerer mange aspekter af MEN1, herunder nye behandlinger til dette tilstand.
Find ud af om kliniske undersøgelser passer til dig.
Hvilke kliniske studier for MEN1 søger deltagere?
Du kan se en filtreret liste over kliniske studier af MEN1, der er åbne og rekrutterer på
www.ClinicalTrials.gov. Du kan udvide eller indsnævre listen til at omfatte kliniske studier fra industri, universiteter og enkeltpersoner; National Institutes of Health gennemgår imidlertid ikke disse undersøgelser og kan ikke sikre, at de er sikre. Tal altid med din sundhedsudbyder, inden du deltager i et klinisk studie.