O gene p53, como o gene Rb, é um gene supressor de tumor, ou seja, sua atividade impede a formação de tumores. Se uma pessoa herda apenas uma cópia funcional do gene p53 de seus pais, ela está predisposta ao câncer e geralmente desenvolve vários tumores independentes em uma variedade de tecidos no início da idade adulta. Essa condição é rara e é conhecida como síndrome de Li-Fraumeni. No entanto, as mutações no p53 são encontradas na maioria dos tipos de tumor e, portanto, contribuem para a complexa rede de eventos moleculares que levam à formação do tumor.
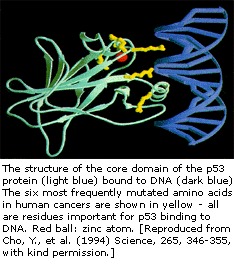
O gene p53 foi mapeado no cromossomo 17. Na célula, a proteína p53 liga-se ao DNA, que por sua vez estimula outro gene a produzir uma proteína chamada p21, que interage com uma proteína estimuladora da divisão celular (cdk2). Quando p21 é complexado com cdk2, a célula não pode passar para o próximo estágio de divisão celular. O p53 mutante não consegue mais se ligar ao DNA de maneira eficaz e, como consequência, a proteína p21 não é disponibilizada para atuar como o “sinal de parada” da divisão celular. Assim, as células se dividem incontrolavelmente e formam tumores.
A ajuda para desvendar os mecanismos moleculares do crescimento canceroso veio do uso de camundongos como modelos para o câncer humano, nos quais poderosas técnicas de “eliminação do gene” podem ser usadas. A quantidade de informações que existe sobre todos os aspectos da função normal do p53 e da expressão mutante em cânceres humanos é agora vasta, refletindo seu papel fundamental na patogênese dos cânceres humanos. É claro que o p53 é apenas um componente de uma rede de eventos que culminam na formação do tumor.