Het p53-gen, zoals het Rb-gen, is een tumorsuppressorgen, dwz zijn activiteit stopt de vorming van tumoren. Als een persoon slechts één functionele kopie van het p53-gen van zijn ouders erft, is hij vatbaar voor kanker en ontwikkelt hij in de vroege volwassenheid gewoonlijk verschillende onafhankelijke tumoren in verschillende weefsels. Deze aandoening is zeldzaam en staat bekend als het Li-Fraumeni-syndroom. Mutaties in p53 worden echter in de meeste tumortypen aangetroffen en dragen zo bij aan het complexe netwerk van moleculaire gebeurtenissen die tot tumorvorming leiden.
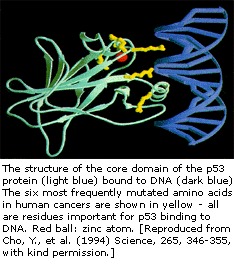
Het p53-gen is in kaart gebracht op chromosoom 17. In de cel, p53-eiwit bindt DNA, dat op zijn beurt een ander gen stimuleert om een eiwit genaamd p21 te produceren dat een interactie aangaat met een celdelingsstimulerend eiwit (cdk2). Wanneer p21 gecomplexeerd is met cdk2, kan de cel niet doorgaan naar het volgende stadium van celdeling. Mutant p53 kan DNA niet langer op een effectieve manier binden, en als gevolg hiervan wordt het p21-eiwit niet beschikbaar gemaakt om te dienen als het “stopsignaal” voor celdeling. Cellen delen zich dus oncontroleerbaar en vormen tumoren.
Hulp bij het ontrafelen van de moleculaire mechanismen van kankergroei is afkomstig van het gebruik van muizen als modellen voor kanker bij de mens, waarbij krachtige “gen-knock-out” -technieken kunnen worden gebruikt. De hoeveelheid informatie die bestaat over alle aspecten van de normale functie van p53 en de expressie van mutanten bij menselijke kankers is nu enorm, wat de sleutelrol in de pathogenese van menselijke kankers weerspiegelt. Het is duidelijk dat p53 slechts één onderdeel is van een netwerk van gebeurtenissen die uitmonden in tumorvorming.