Das p53-Gen wie das Rb-Gen, ist ein Tumorsuppressorgen, dh seine Aktivität stoppt die Bildung von Tumoren. Wenn eine Person nur eine funktionelle Kopie des p53-Gens von ihren Eltern erbt, ist sie für Krebs prädisponiert und entwickelt im frühen Erwachsenenalter normalerweise mehrere unabhängige Tumoren in einer Vielzahl von Geweben. Dieser Zustand ist selten und als Li-Fraumeni-Syndrom bekannt. Mutationen in p53 werden jedoch in den meisten Tumortypen gefunden und tragen so zum komplexen Netzwerk molekularer Ereignisse bei, die zur Tumorbildung führen.
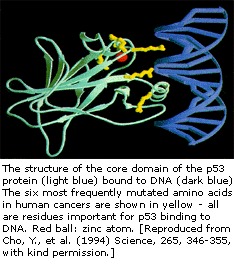
Das p53-Gen wurde auf Chromosom 17 abgebildet. In der Zelle p53-Protein bindet DNA, die wiederum ein anderes Gen stimuliert, um ein Protein namens p21 zu produzieren, das mit einem zellteilungsstimulierenden Protein (cdk2) interagiert. Wenn p21 mit cdk2 komplexiert ist, kann die Zelle nicht zur nächsten Stufe der Zellteilung übergehen. Die Mutante p53 kann DNA nicht mehr effektiv binden, und infolgedessen wird das p21-Protein nicht als „Stoppsignal“ für die Zellteilung zur Verfügung gestellt. Auf diese Weise teilen sich Zellen unkontrolliert und bilden Tumore.
Die Aufklärung der molekularen Mechanismen des Krebswachstums wurde durch die Verwendung von Mäusen als Modelle für Krebs beim Menschen ermöglicht, bei denen leistungsstarke „Gen-Knockout“ -Techniken eingesetzt werden können. Die Informationsmenge, die zu allen Aspekten der normalen Funktion von p53 und der Mutantenexpression bei Krebserkrankungen beim Menschen vorliegt, ist jetzt riesig und spiegelt seine Schlüsselrolle bei der Pathogenese von Krebserkrankungen beim Menschen wider. Es ist klar, dass p53 nur eine Komponente eines Netzwerks von Ereignissen ist, die zur Tumorbildung führen.