El gen p53, como el gen Rb, es un gen supresor de tumores, es decir, su actividad detiene la formación de tumores. Si una persona hereda solo una copia funcional del gen p53 de sus padres, está predispuesta al cáncer y generalmente desarrolla varios tumores independientes en una variedad de tejidos en la edad adulta temprana. Esta condición es rara y se conoce como síndrome de Li-Fraumeni. Sin embargo, las mutaciones en p53 se encuentran en la mayoría de los tipos de tumores y, por lo tanto, contribuyen a la compleja red de eventos moleculares que conducen a la formación de tumores.
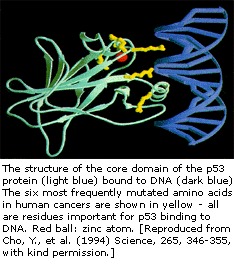
El gen p53 se ha mapeado en el cromosoma 17. En la célula, la proteína p53 se une al ADN, que a su vez estimula a otro gen para producir una proteína llamada p21 que interactúa con una proteína que estimula la división celular (cdk2). Cuando p21 forma un complejo con cdk2, la célula no puede pasar a la siguiente etapa de división celular. La p53 mutante ya no puede unirse al ADN de forma eficaz y, como consecuencia, la proteína p21 no está disponible para actuar como «señal de parada» para la división celular. Por lo tanto, las células se dividen sin control y forman tumores.
La ayuda para desentrañar los mecanismos moleculares del crecimiento canceroso proviene del uso de ratones como modelos para el cáncer humano, en los que se pueden utilizar potentes técnicas de «eliminación genética». La cantidad de información que existe sobre todos los aspectos de la función normal de p53 y la expresión de mutantes en los cánceres humanos es ahora enorme, lo que refleja su papel clave en la patogénesis de los cánceres humanos. Está claro que p53 es solo un componente de una red de eventos que culminan en la formación de tumores.